Multiplex Droplet Digital PCR for ctDNA Analysis: A Comprehensive Guide for Advanced Cancer Research and Biomarker Development
This article provides a comprehensive exploration of multiplex droplet digital PCR (ddPCR) for circulating tumor DNA (ctDNA) analysis, a transformative technology in precision oncology.
Multiplex Droplet Digital PCR for ctDNA Analysis: A Comprehensive Guide for Advanced Cancer Research and Biomarker Development
Abstract
This article provides a comprehensive exploration of multiplex droplet digital PCR (ddPCR) for circulating tumor DNA (ctDNA) analysis, a transformative technology in precision oncology. Tailored for researchers, scientists, and drug development professionals, it covers foundational principles, from ctDNA biology and ddPCR partitioning to the strategic advantage of multiplexing over singleplex assays. The content details methodological workflows for assay design, sample processing, and data analysis, alongside diverse applications in cancer detection, minimal residual disease (MRD) monitoring, and treatment response assessment. It further addresses critical troubleshooting and optimization strategies to ensure high sensitivity and low false-positive rates, and concludes with rigorous validation frameworks and comparative analyses with other genomic technologies like NGS and qPCR.
The Fundamentals of ctDNA and Multiplex ddPCR: Principles and Core Technologies
Biology and Origin of Circulating Tumor DNA
Circulating tumor DNA (ctDNA) refers to fragmented DNA molecules released from tumor cells into the bloodstream and other bodily fluids. These fragments carry tumor-specific genetic and epigenetic alterations, providing a non-invasive means to interrogate the tumor genome.
Biological Characteristics and Release Mechanisms
CtDNA release occurs through multiple mechanisms, primarily via apoptosis, necrosis, and active secretion from tumor cells [1] [2].
- Apoptosis: Programmed cell death produces ctDNA fragments with a characteristic ladder-like pattern, with a peak fragment size of approximately 167 base pairs [1]. This size corresponds to DNA wrapped around a single nucleosome (147 bp) plus linker DNA, protected from cleavage by histones [1]. Caspase-activated DNase (CAD) and other nucleases execute systematic DNA fragmentation during apoptosis [1].
- Necrosis: Uncontrolled cell death results in random DNA fragmentation, generating larger DNA fragments of up to many kilobase pairs [1]. Necrotic tumor cells release various immune cell attractants and are eliminated by macrophages, leading to digested ctDNA release into circulation [1].
- Active Secretion: Viable tumor cells can actively release DNA through extracellular vesicles (EVs) or other mechanisms, though this process is less well-characterized [3].
CtDNA exists as either single- or double-stranded DNA in plasma or serum, typically shorter than non-tumor cell-free DNA (cfDNA) [2]. In cancer patients, ctDNA represents a small fraction (0.1% to 90%) of total cell-free DNA, with proportion increasing with tumor burden [4].
Fragment Properties and Clearance
CtDNA exhibits distinct fragmentation patterns compared to non-tumor cfDNA. Research indicates ctDNA fragments are typically shorter (20-50 base pairs) than cfDNA from healthy cells [5]. The half-life of ctDNA in circulation is short, estimated between 16 minutes to several hours, enabling real-time monitoring of tumor dynamics [4].
Table 1: Biological Properties of ctDNA
| Characteristic | Description | Clinical Significance |
|---|---|---|
| Primary Sources | Apoptosis, necrosis, active secretion | Indicates tumor cell turnover and treatment response |
| Typical Fragment Size | ~167 bp (apoptosis); variable (necrosis) | Helps distinguish tumor-derived from normal cfDNA |
| Circulation Half-life | 16 minutes to several hours | Enables real-time monitoring of tumor dynamics |
| Percentage of Total cfDNA | 0.1% in early-stage to >90% in late-stage disease | Correlates with tumor burden |
Clinical Significance and Applications
CtDNA analysis has emerged as a transformative approach in oncology, with applications spanning diagnosis, monitoring, and treatment selection.
Diagnostic and Prognostic Utility
CtDNA carries tumor-specific molecular alterations including point mutations, copy number variations, chromosomal rearrangements, and methylation pattern changes [2]. These characteristics enable non-invasive cancer detection and molecular profiling.
DNA methylation changes are particularly valuable biomarkers as they occur early in carcinogenesis and are highly recurrent across tumor types [6] [7]. Methylation patterns can distinguish cancer types while common methylation patterns allow multi-cancer detection [7].
CtDNA levels correlate with tumor stage and burden. Patients with metastatic disease demonstrate significantly higher ctDNA levels than those with localized cancers [3]. In non-small cell lung cancer, detection rates range from 38.7-46.8% in non-metastatic disease to 70.2-83.0% in metastatic cases [6].
Monitoring Treatment Response and Resistance
The short half-life of ctDNA makes it ideal for monitoring treatment response. Changes in ctDNA levels can precede radiographic evidence of response or progression [4].
- Molecular response: CtDNA clearance after treatment correlates with improved outcomes
- Resistance monitoring: Emerging mutations can identify acquired resistance to targeted therapies
- Minimal Residual Disease (MRD): Detection of ctDNA after curative-intent surgery predicts recurrence [4]
Longitudinal ctDNA monitoring enables dynamic assessment of tumor evolution and treatment efficacy, potentially guiding therapy modifications before clinical progression becomes evident [4].
Technical Considerations and Challenges
Despite its promise, ctDNA analysis faces several challenges:
- Low abundance: Especially in early-stage disease or low-shedding tumors
- Lack of standardization: Preanalytical variables (blood collection, processing, storage) and analytical methods affect results
- Sensitivity limitations: Current assays may miss very low-frequency variants
- Clonal hematopoiesis: Age-related mutations in blood cells can confound interpretation
Table 2: Clinical Applications of ctDNA Analysis
| Application | Utility | Current Status |
|---|---|---|
| Early Cancer Detection | Identify cancer before symptomatic presentation | Emerging; multi-cancer detection tests in development |
| Treatment Selection | Identify targetable mutations without invasive biopsy | FDA-approved tests available (e.g., for EGFR mutations in NSCLC) |
| MRD Detection | Identify residual disease after curative-intent treatment | Clinical validation ongoing; prognostic value established |
| Treatment Monitoring | Assess response and emergence of resistance | Growing evidence supporting clinical utility |
| Prognostication | Predict outcomes based on ctDNA levels | Established correlation with survival in multiple cancers |
Experimental Approaches and Methodologies
Detection Technologies
CtDNA detection requires highly sensitive methods due to its low abundance in total cfDNA. Current approaches include:
- PCR-based methods: Digital PCR (dPCR), droplet digital PCR (ddPCR), and BEAMing enable highly sensitive detection of known mutations with rapid turnaround times [8] [4]. These methods are ideal for tracking specific mutations during treatment.
- Next-generation sequencing (NGS): Targeted panels, whole-exome, and whole-genome sequencing provide comprehensive mutation profiling [8] [4]. Unique molecular identifiers (UMIs) help distinguish true mutations from sequencing artifacts [4].
- Methylation analysis: Bisulfite conversion followed by sequencing or PCR detects cancer-specific methylation patterns [8] [6]. Both whole-genome and targeted approaches are used.
- Fragmentomics: Analysis of cfDNA fragmentation patterns, sizes, and end characteristics provides an emerging approach to distinguish ctDNA from normal cfDNA [8].
Multiplex ddPCR for Methylation-Based Detection
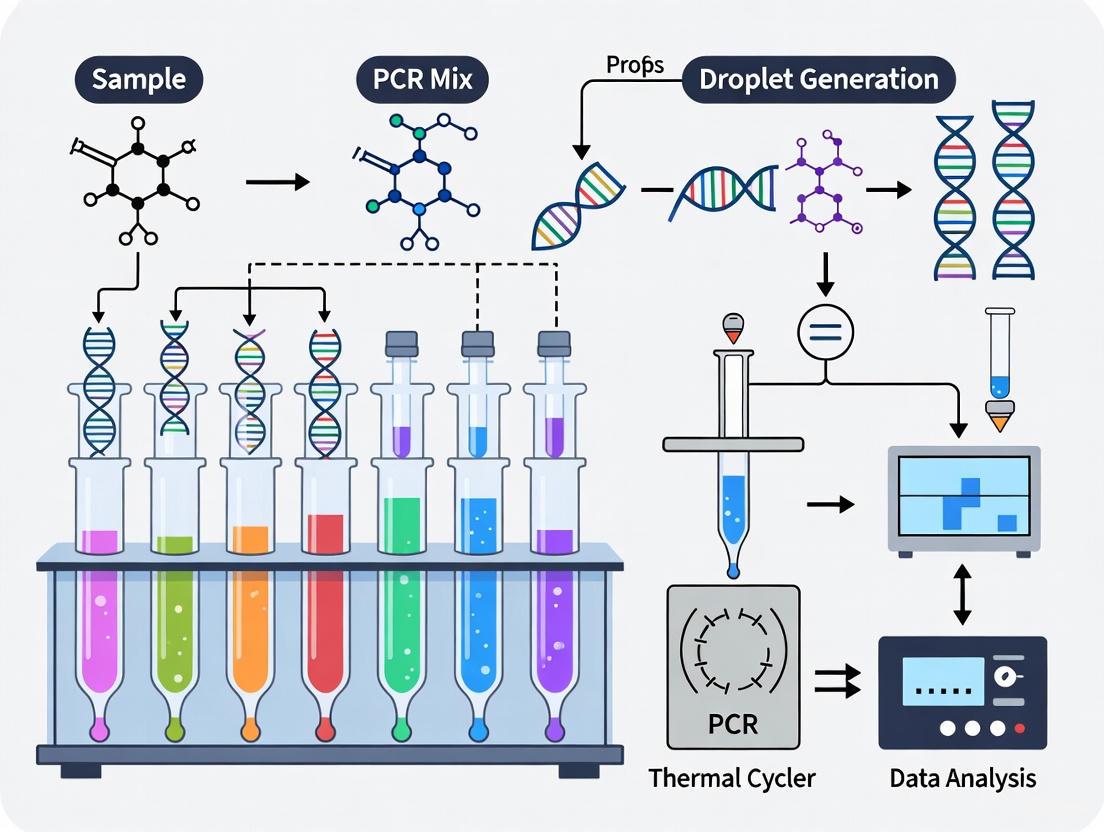
Droplet digital PCR has emerged as a powerful platform for ctDNA detection due to its high sensitivity, absolute quantification, and compatibility with multiplex assays. A representative workflow for multiplex ddPCR methylation analysis includes:
Detailed Protocol: Multiplex Methylation-Specific ddPCR [6] [7]
Sample Collection and Processing:
- Collect whole blood in EDTA tubes (e.g., 9 mL)
- Process within 4 hours of venipuncture
- Centrifuge at 2,000 × g for 10 minutes to separate plasma
- Aliquot and store plasma at -80°C until analysis
cfDNA Extraction:
- Thaw plasma samples at 5°C
- Centrifuge at 10,000 × g for 10 minutes to remove debris
- Add spike-in control DNA (e.g., ~9,000 copies/mL CPP1) for extraction efficiency monitoring
- Extract cfDNA using commercial kits (e.g., QIAsymphony DSP Circulating DNA Kit)
- Elute in appropriate buffer (e.g., 60 μL Plasma Elution Buffer)
Bisulfite Conversion:
- Concentrate extracted DNA to 20 μL using centrifugal filters
- Perform bisulfite conversion using commercial kits (e.g., Zymo Research EZ DNA Methylation-Lightning Kit)
- Elute bisulfite-converted DNA in 15 μL M-Elution Buffer
- Use converted DNA within 10 days
Multiplex ddPCR Assay:
- Design assays targeting differentially methylated regions identified through bioinformatics analysis
- Select 3-5 methylation markers for multiplexing to increase sensitivity
- Set up ddPCR reaction mixture according to manufacturer's protocol
- Include no-template controls and positive controls (e.g., cancer cell line DNA)
- Run ddPCR with appropriate thermal cycling conditions
- Analyze droplets using automated droplet readers
Data Analysis and Interpretation:
- Calculate methylation ratios for each target
- Apply predetermined cut-off values for ctDNA positivity
- Normalize results using control assays
- Determine ctDNA status based on combined read-out from multiple targets
Research Reagent Solutions
Table 3: Essential Research Reagents for ctDNA Analysis
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Blood Collection Tubes | EDTA tubes | Prevents coagulation and preserves cfDNA |
| cfDNA Extraction Kits | QIAsymphony DSP Circulating DNA Kit (Qiagen) | Isolves cell-free DNA from plasma |
| Bisulfite Conversion Kits | EZ DNA Methylation-Lightning Kit (Zymo Research) | Converts unmethylated cytosines to uracils |
| ddPCR Supermixes | ddPCR Supermix for Probes (Bio-Rad) | Enables droplet digital PCR reactions |
| Methylation-Specific Probes | FAM/HEX-labeled probes for target genes | Detects methylated alleles in bisulfite-converted DNA |
| Quality Control Assays | EMC7 65bp/250bp assays, immunoglobulin gene assays | Assesses total cfDNA concentration and contamination |
CtDNA represents a transformative biomarker in oncology, offering insights into tumor biology, dynamics, and heterogeneity through minimally invasive liquid biopsies. Understanding its biological origins, release mechanisms, and clinical significance enables researchers to develop increasingly sophisticated applications for cancer detection, monitoring, and personalized treatment. Multiplex ddPCR approaches, particularly those leveraging DNA methylation biomarkers, provide sensitive, cost-effective platforms for ctDNA analysis across diverse cancer types and clinical scenarios. As technologies advance and standardization improves, ctDNA analysis is poised to become an integral component of precision oncology, enabling more dynamic and comprehensive cancer management.
Droplet Digital PCR (ddPCR) represents a transformative approach in nucleic acid quantification, enabling absolute target measurement without standard curves. This technology is particularly vital in circulating tumor DNA (ctDNA) analysis for cancer research, where detecting rare mutations against a wild-type background demands exceptional sensitivity and precision [9]. The core innovation of ddPCR lies in its partitioning of samples into thousands of nanoliter-sized droplets, functioning as independent PCR microreactors. This partitioning facilitates binary endpoint detection that enables absolute quantification through Poisson statistics [10] [11]. For researchers and drug development professionals investigating ctDNA, ddPCR provides the necessary technical capabilities to address challenges such as low ctDNA fraction in total cell-free DNA, sometimes less than 0.01%, and the need for precise longitudinal monitoring of treatment response [9] [6]. This application note details the core mechanics, experimental protocols, and research applications of ddPCR to support its implementation in ctDNA research workflows.
Fundamental Principles of ddPCR
The Partitioning Process and Endpoint Detection
The ddPCR workflow fundamentally differs from quantitative real-time PCR (qPCR) through its initial partitioning step. In ddPCR, each sample is partitioned into approximately 20,000 nanoliter-sized water-in-oil droplets, where each droplet acts as an individual PCR microreactor [12]. This massive partitioning occurs before amplification, randomly distributing target DNA molecules across the droplets according to Poisson statistics. Following partitioning, conventional PCR amplification occurs within each droplet with temperature cycling that includes denaturation, primer hybridization, and elongation [10].
Critically, ddPCR utilizes endpoint detection rather than monitoring amplification in real-time. After PCR amplification is complete, each droplet is analyzed for fluorescence signal to determine whether it contains amplified target DNA (positive) or not (negative) [10] [11]. This binary readout system converts the continuous analog measurement of traditional PCR into a digital format of ones and zeros, hence the "digital" designation. The fraction of positive droplets, determined by this endpoint measurement, is then used to absolutely quantify the original target concentration in the sample [11].
Absolute Quantification via Poisson Statistics
Absolute quantification in ddPCR relies on the mathematical foundation of Poisson statistics, which describes the probability of target molecule distribution across partitions when that distribution is random [10]. The core principle states that the average number of target molecules per droplet (λ) can be calculated from the proportion of negative droplets using the formula: λ = -ln(1-p), where p represents the fraction of positive droplets [10] [11].
The confidence in quantification depends significantly on the number of partitions analyzed. With approximately 20,000 droplets, optimal precision is achieved when about 20% of droplets are positive (λ ≈ 1.6), providing the ideal balance between empty and saturated partitions for statistical confidence [10] [11]. This statistical foundation enables ddPCR to provide absolute quantification without standard curves, eliminating concerns about amplification efficiency variations that can affect qPCR results [10].
Table 1: Key Performance Advantages of ddPCR in ctDNA Research
| Feature | Technical Advantage | Impact on ctDNA Analysis |
|---|---|---|
| Partitioning Technology | Divides sample into ~20,000 nanoliter droplets [12] | Enables detection of rare mutations present at very low frequencies [9] |
| Absolute Quantification | No standard curves required; uses Poisson statistics [10] [11] | Eliminates calibration variability for consistent longitudinal monitoring [9] |
| Enhanced Sensitivity | Can detect minority clones at frequencies as low as 0.1-1% [13] | Crucial for detecting low fractional abundance ctDNA in total cfDNA [9] [6] |
| Tolerance to Inhibitors | Sample partitioning reduces effect of PCR inhibitors [12] [10] | Improves reliability with complex biological samples like plasma [6] |
| Precision at Low Concentrations | Superior accuracy for low-copy nucleic acids [12] | Essential for monitoring minimal residual disease and early recurrence [6] |
ddPCR Workflow and Protocol
The standard ddPCR protocol involves sample preparation, partitioning, amplification, and droplet reading phases. The following workflow diagram illustrates this process:
Detailed Experimental Protocol for ctDNA Analysis
The following protocol is adapted from validated methodologies for ctDNA detection in cancer research [9] [6]:
Sample Preparation and DNA Extraction
- Plasma Separation: Collect whole blood in EDTA tubes and centrifuge at 2,000 × g for 10 minutes within 4 hours of venipuncture to isolate plasma [6].
- cfDNA Extraction: Use the QIAsymphony SP system with DSP Circulating DNA Kit or similar. Elute cfDNA in 50-60 μL of elution buffer [6].
- DNA Quantification: Assess total cfDNA concentration using a reference assay (e.g., EMC7 65 bp amplicon) and check for high-molecular-weight DNA contamination with a longer amplicon assay (e.g., EMC7 250 bp) [6].
- Bisulfite Conversion: For methylation analysis, concentrate extracted DNA to 20 μL and treat with bisulfite conversion kit (e.g., EZ DNA Methylation-Lightning Kit). Elute converted DNA in 15 μL M-Elution Buffer [6].
ddPCR Reaction Setup
- Reaction Composition:
- 10-20 μL of sample DNA
- 1× ddPCR Supermix
- Target-specific primers and probes (optimized concentrations, typically 250-900 nM each)
- Nuclease-free water to final volume (typically 20-22 μL before partitioning) [6]
- Optimization Notes:
- Annealing/extension temperature and oligonucleotide concentrations significantly impact assay performance and "rain" minimization [14].
- Include positive and negative controls in each run.
Droplet Generation and PCR Amplification
- Partitioning: Transfer reaction mix to DG8 Cartridge and generate droplets using Droplet Generator. Approximately 20,000 droplets per sample should be produced [12].
- PCR Amplification: Transfer droplets to 96-well plate and seal. Perform amplification with thermal cycling conditions optimized for the target:
- Endpoint Hold: 4°C or 12°C until droplet reading
Droplet Reading and Data Analysis
- Droplet Reading: Transfer plate to Droplet Reader which measures fluorescence in each droplet.
- Threshold Setting: Set fluorescence thresholds to distinguish positive and negative droplet populations. Use objective separation algorithms when possible to minimize "rain" (intermediate signals) [14].
- Concentration Calculation: Use Poisson statistics to calculate absolute copies/μL of input sample: Concentration = -ln(1 - (positive droplets/total droplets)) × (total partitions/reaction volume) [10].
Essential Research Reagents and Materials
Successful implementation of ddPCR for ctDNA research requires carefully selected reagents and optimization. The following table details essential components:
Table 2: Research Reagent Solutions for ddPCR in ctDNA Analysis
| Reagent/Material | Function | Application Notes |
|---|---|---|
| ddPCR Supermix | Provides optimized buffer, enzymes, and dNTPs for amplification | Select probe-based or EvaGreen formats based on assay design; crucial for droplet stability [6] |
| Target-Specific Assays | Primers and probes for target detection | Optimize concentrations (250-900 nM); validated assays reduce optimization time [14] [6] |
| Bisulfite Conversion Kits | Converts unmethylated cytosines to uracils for methylation analysis | Essential for methylation-specific ddPCR; conversion efficiency affects results [6] |
| Droplet Generation Oil | Creates stable water-in-oil emulsions | Formulation critical for droplet integrity during thermal cycling [12] |
| cfDNA Extraction Kits | Isolves cell-free DNA from plasma | High recovery rates essential for low-abundance ctDNA; includes carrier DNA or spike-ins [6] |
| Quality Control Assays | Assesses sample quality and quantity | Include reference gene assays, extraction controls, and genomic DNA contamination checks [6] |
Application in ctDNA Research: A Case Protocol
The following case example demonstrates ddPCR application in lung cancer ctDNA detection using methylation markers:
Multiplexed Methylation-Specific ddPCR
Recent research has established multiplexed methylation-specific ddPCR assays for lung cancer detection [6]. This protocol enables simultaneous detection of five tumour-specific methylation markers, significantly enhancing detection sensitivity compared to single-plex assays.
Experimental Workflow:
- Marker Selection: Identify differentially methylated regions (DMRs) through bioinformatics analysis of public methylation arrays (e.g., TCGA data). Select DMRs with mean beta-value differences >0.5 between tumor and normal samples [6].
- Assay Design: Design primers and probes targeting the selected DMRs, with emphasis on CpG islands within promoter regions.
- Multiplex Optimization: Systematically optimize annealing temperatures and primer concentrations to achieve balanced amplification of all five targets while minimizing "rain" [14] [6].
- Validation: Test assay performance in training cohorts of healthy controls and patients with confirmed lung cancer.
- Clinical Application: Apply validated multiplex to plasma samples from patients with non-metastatic and metastatic disease.
Performance Metrics: In validation studies, this approach demonstrated ctDNA-positive rates of 38.7-46.8% in non-metastatic disease and 70.2-83.0% in metastatic cases, highlighting both the power and limitations of current ctDNA detection technologies [6].
The statistical principles underlying this quantification method are visualized below:
Droplet Digital PCR represents a powerful analytical platform that leverages partitioning, endpoint PCR, and Poisson-based absolute quantification to address significant challenges in molecular analysis. For ctDNA research in particular, these core mechanics enable the detection and precise quantification of rare mutant alleles in complex biological samples, providing researchers and drug development professionals with a robust tool for cancer monitoring, treatment response assessment, and minimal residual disease detection. The continued refinement of ddPCR technologies, including enhanced multiplexing capabilities and improved workflow efficiency, promises to further expand its utility in both basic research and clinical applications.
Circulating tumour DNA (ctDNA) analysis has emerged as a non-invasive tool for cancer management, enabling applications from early detection to monitoring treatment response [6]. A significant challenge in this field is the low abundance of ctDNA in plasma, especially in early-stage disease or minimal residual disease (MRD) settings [15]. Droplet digital PCR (ddPCR) provides the sensitivity required for ctDNA detection, but single-target approaches may miss tumour-derived DNA due to tumour heterogeneity or low variant allele frequencies [7].
Multiplex ddPCR addresses this limitation by simultaneously analysing multiple biomarkers within a single reaction, significantly enhancing assay sensitivity without compromising specificity [6]. This application note details the development, validation, and implementation of multiplex ddPCR assays for ctDNA analysis, providing researchers with structured protocols, performance data, and practical implementation strategies to leverage the multiplexing advantage in cancer research.
Performance Comparison: Single-Target vs. Multi-Target Approaches
Empirical studies across multiple cancer types demonstrate that multiplex approaches consistently outperform single-target assays by increasing the probability of detecting low-abundance ctDNA fragments.
Table 1: Comparative Performance of Single-Target vs. Multi-Target ddPCR Assays in Cancer Detection
| Cancer Type | Assay Type | Number of Targets | Sensitivity | Specificity | Reference Application |
|---|---|---|---|---|---|
| Lung Cancer | Methylation-specific ddPCR | 5 | 38.7-46.8% (non-metastatic); 70.2-83.0% (metastatic) | Not specified | Early detection and monitoring [6] |
| Multi-Cancer | Methylation ddPCR | 3 | 53.8-100% (varies by cancer type) | 80-100% (varies by cancer type) | Detection of eight cancer types [7] |
| Colorectal Cancer | Tumour-informed ddPCR | 1 vs. 16 | 72/92 (ST) vs. 88/92 (MT) preoperatively | Similar in postoperative samples | Postoperative risk stratification [15] |
The performance gains are particularly evident in challenging clinical scenarios. In lung cancer, a 5-plex methylation-specific ddPCR assay demonstrated substantially higher detection rates in metastatic disease (70.2-83.0%) compared to non-metastatic cases (38.7-46.8%), with sensitivity variations observed across histological subtypes [6]. For multi-cancer detection, a three-target methylation ddPCR assay achieved remarkable accuracy (cross-validated AUC: 0.948) across eight different cancer types, though performance varied by cancer type [7].
Not all studies show dramatic advantages for multiplex approaches. In colorectal cancer postoperative monitoring, a comparison between single-target ddPCR and a 16-plex NGS assay (Signatera) showed similar performance in longitudinal monitoring, suggesting that context and application influence the optimal degree of multiplexing [15].
Experimental Protocol: Developing a Multiplex Methylation ddPCR Assay
This protocol details the development of a 5-plex methylation-specific ddPCR assay for lung cancer detection, adaptable to other cancer types [6].
Biomarker Discovery and Selection
- Bioinformatic Analysis: Identify differentially methylated CpG sites through analysis of public methylation arrays (e.g., TCGA data).
- Selection Criteria: Choose CpGs with mean beta-value differences >0.5 between tumour and normal samples, located within CpG islands.
- Feature Reduction: Apply recursive feature elimination (RFE) with 10-fold cross-validation to identify the most discriminatory markers.
- Literature Integration: Include markers with prior evidence (e.g., HOXA9 in lung cancer) [6].
Sample Processing and DNA Extraction
- Blood Collection: Draw blood into EDTA or Cell-Free DNA BCT tubes.
- Plasma Isolation: Centrifuge within 4 hours of venepuncture at 2,000 × g for 10 minutes.
- cfDNA Extraction: Extract from 4 mL plasma using the QIAsymphony DSP Circulating DNA Kit or similar.
- Quality Assessment:
Bisulfite Conversion and ddPCR Setup
- Concentration: Concentrate extracted DNA to 20 μL using Amicon Ultra-0.5 Centrifugal Filter units.
- Bisulfite Conversion: Use the EZ DNA Methylation-Lightning Kit with elution in 15 μL M-Elution Buffer.
- Reaction Setup:
- Prepare 22 μL reactions with 11 μL of 2× ddPCR Supermix for Probes.
- Add optimized primer and probe concentrations (determined during validation).
- Include bisulfite-converted DNA (volume dependent on input cfDNA).
- Droplet Generation: Use the QX200 AutoDG Droplet Generator.
- PCR Amplification:
- 95°C for 10 minutes (enzyme activation)
- 40 cycles of: 94°C for 30 seconds, annealing temperature (optimized) for 60 seconds
- 98°C for 10 minutes (enzyme deactivation)
- 4°C hold
- Droplet Reading: Analyze on QX200 Droplet Reader [6].
Data Analysis
- Quality Control:
- Assess extraction efficiency via spike-in recovery.
- Evaluate sample quality using EMC7 65/250 ratio.
- Droplet Classification: Use cluster identification algorithms (e.g., kernel density estimation, Gaussian mixture models) [17].
- ctDNA Status Determination: Apply validated cut-off methods for calling samples positive [6].
Multiplexing Strategies and Technical Optimization
Effective multiplex ddPCR requires strategic assay design and careful optimization to overcome technical challenges associated with multiple primer-probe combinations.
Multiplexing Strategies for Two-Color Systems
Table 2: Multiplexing Strategies for Two-Color ddPCR Systems
| Strategy | Principle | Application Example | Key Considerations |
|---|---|---|---|
| Probe Mixing | Target detected by both FAM and HEX probes generates combined fluorescent signal | Detection of 4 PIK3CA mutations [18] | Requires careful probe concentration optimization |
| Amplitude-Based Multiplexing | Different targets with same fluorophore distinguished by fluorescence amplitude | 4-plex detection of Vibrio parahaemolyticus genes [18] | Maintain sufficient concentration difference between probes |
| Combined Approach | Integration of both probe mixing and amplitude-based strategies | 5-plex detection of biothreat pathogens [18] | Maximum multiplexing in standard systems |
Critical Optimization Parameters
Oligonucleotide Concentrations:
- Test primer concentrations (typically 400-900 nM) and probe concentrations (150-250 nM)
- Utilize "experience matrix" to evaluate multiple parameter combinations [19]
Thermal Cycling Conditions:
- Optimize annealing/extension temperature using gradient PCR
- Balance amplification efficiency across all targets
Rain Reduction:
- Optimize conditions to minimize intermediate fluorescence signals
- Use separation value calculation considering fluorescence distance and variation [19]
False Positive Management:
- Include extensive negative controls
- Establish threshold values for mutation calling [16]
Essential Reagents and Research Tools
Table 3: Research Reagent Solutions for Multiplex ddPCR
| Reagent/Tool | Function | Examples/Specifications |
|---|---|---|
| Nucleic Acid Extraction Kits | Isolation of high-quality cfDNA from plasma | QIAsymphony DSP Circulating DNA Kit, QIAamp Circulating Nucleic Acid Kit [6] [16] |
| Bisulfite Conversion Kits | Conversion of unmethylated cytosines to uracils for methylation analysis | EZ DNA Methylation-Lightning Kit [6] [7] |
| ddPCR Master Mix | Provides optimal environment for partitioned PCR | ddPCR Supermix for Probes (no dUTP) [6] [16] |
| Fluorescent Probes | Sequence-specific detection with different fluorophores | FAM and HEX-labeled TaqMan probes, optionally with LNA modifications [16] [19] |
| Droplet Generation Oil | Creates stable water-in-oil emulsion for partitioning | DG Cartridges and Droplet Generation Oil [16] |
| Quality Control Assays | Assessment of extraction efficiency and sample quality | Exogenous spike-ins (CPP1, XenT), reference gene assays (RPP30, EMC7) [6] [16] |
| Analysis Software | Automated droplet classification and quantification | QuantaSoft, ddpcr R package [17] |
Data Analysis and Interpretation
Robust data analysis is crucial for accurate interpretation of multiplex ddPCR results, particularly given the complex fluorescence patterns generated.
Droplet Classification and Rain Management
The ddpcr R package provides improved cluster identification through kernel density estimation and Gaussian mixture models, offering advantages over proprietary software, especially for challenging samples like FFPE-derived DNA [17]. The package includes:
- Automated well failure identification
- Outlier droplet exclusion
- Empty droplet identification
- Cluster assignment with rain management
Quantification and Statistical Analysis
Absolute Quantification: Calculate template concentrations using Poisson distribution applied to positive and negative droplet counts [20].
Multiplexing Benefit Assessment:
- Compare sensitivity and specificity to single-target approaches
- Evaluate statistical significance using appropriate tests (e.g., Wilcoxon matched-pairs signed rank test) [20]
Longitudinal Monitoring: Track ctDNA dynamics across multiple timepoints to assess treatment response and disease progression [6].
Multiplex ddPCR represents a significant advancement in ctDNA analysis, offering enhanced sensitivity while maintaining specificity through simultaneous detection of multiple biomarkers. The structured protocols and optimization strategies presented here provide researchers with a framework for implementing this powerful technology in cancer research. As the field progresses, standardized multiplex assays will play an increasingly important role in translational cancer research, potentially bridging the gap between liquid biopsy development and clinical application.
Circulating tumor DNA (ctDNA) has emerged as a pivotal biomarker in liquid biopsies, enabling non-invasive monitoring of tumor dynamics and treatment response in cancer patients [4]. This fragmented DNA, released into the bloodstream by tumor cells, carries tumor-specific genomic alterations that provide a comprehensive representation of tumor heterogeneity [4]. The analysis of ctDNA is particularly valuable for longitudinal monitoring of disease burden, assessment of minimal residual disease (MRD), and early detection of emergent resistance mutations during therapy [4] [16].
Among the various technological platforms available for ctDNA analysis, droplet digital PCR (ddPCR) offers distinct advantages for precise, absolute quantification of target DNA molecules with exceptional sensitivity and specificity [7] [16]. The recent development of multiplex ddPCR assays further enhances this approach by enabling simultaneous detection of multiple biomarker types within a single reaction, conserving precious patient samples while providing comprehensive molecular profiling [7]. This application note focuses on three principal biomarker categories detectable via multiplex ddPCR: point mutations, DNA methylation patterns, and copy number variations, detailing their clinical significance and optimized detection protocols.
Biomarker Characteristics and Clinical Applications
Table 1: Comparative Analysis of Key ctDNA Biomarkers
| Biomarker Type | Molecular Nature | Detection Challenge | Primary Clinical Utility | Example Genes/Targets |
|---|---|---|---|---|
| Point Mutations | Single nucleotide changes in DNA sequence | Low variant allele frequency (VAF) amidst wild-type DNA | Monitoring tumor burden, tracking resistance mutations, treatment response assessment | KRAS, PIK3CA, EGFR, ESR1 [4] |
| DNA Methylation Patterns | Cytosine methylation in CpG islands | Low abundance of methylated alleles; requires bisulfite conversion | Early cancer detection, tumor origin identification, prognosis assessment | SOX17, CST6, BRMS1 [21]; multi-cancer panels [7] |
| Copy Number Variations (CNVs) | Gains or losses of large genomic regions | Low tumor fraction; background from normal DNA | Detection of oncogene amplification, tumor suppressor loss, genome instability | LINE-1 assays for aneuploidy [22] |
Experimental Protocols
Sample Collection and cfDNA Extraction
Materials:
- Blood collection tubes (Streck Cell-Free DNA BCT, K2 EDTA, or CellSave)
- Maxwell RSC instrument with ccfDNA Plasma Kit (Promega) or QIAamp Circulating Nucleic Acid Kit (Qiagen)
- Qubit Fluorometer 2.0 with dsDNA HS Assay
- Synthetic DNA control (XenT gBlock for extraction efficiency monitoring) [16]
Protocol:
- Blood Collection and Plasma Isolation: Collect peripheral blood into appropriate collection tubes. Process within 4 hours (EDTA) or 96 hours (Streck/CellSave) using a two-step centrifugation: 10 minutes at 1,711 × g at room temperature, followed by 10 minutes at 12,000 × g at 4°C [22].
- cfDNA Extraction: Extract cfDNA from up to 2 mL plasma using validated kits according to manufacturer's protocols. Spike plasma with 20,000 copies of synthetic control DNA prior to extraction to monitor extraction efficiency [16].
- Quantification and Quality Control: Measure cfDNA concentration using fluorometric methods (Qubit). Assess fragment size distribution if possible (typically ~160-170 bp for ctDNA). Store eluates in DNA low-bind tubes at -20°C [16].
Bisulfite Conversion for Methylation Analysis
Materials:
- EZ DNA Methylation Kit (Zymo Research)
- Thermal cycler
- Elution buffer (TE buffer recommended)
Protocol:
- Use 20 ng cfDNA as input for bisulfite conversion according to manufacturer's instructions [7].
- Set elution volume to 2 μL × n + 1 μL, where n represents the number of downstream assays.
- Store bisulfite-converted DNA at -20°C and use within 10 days of conversion to ensure integrity [7].
Multiplex ddPCR Assay Setup
Table 2: Essential Research Reagent Solutions
| Reagent/Category | Specific Product Examples | Function in Workflow |
|---|---|---|
| ddPCR Master Mix | ddPCR SuperMix for Probes (no dUTP), Bio-Rad | Provides optimal reaction environment for partitioned PCR |
| Hydrolysis Probes | PrimeTime LNA probes (FAM/HEX), IDT | Target-specific detection with enhanced discrimination [16] |
| Primers | Custom-designed, Eurogentec | Target-specific amplification |
| Control Templates | gBlocks (IDT), Horizon Reference Standards | Assay validation, limit of detection determination [16] |
| DNA Extraction Kits | QIAamp Circulating Nucleic Acid Kit (Qiagen), ccfDNA Plasma Kit (Promega) | Isolation of high-quality cfDNA from plasma |
| Bisulfite Kits | EZ DNA Methylation Kit (Zymo Research) | Conversion of unmethylated cytosine to uracil for methylation analysis [7] |
Materials:
- QX200 AutoDG Droplet Digital PCR System (Bio-Rad)
- PrimeTime LNA probes with FAM/HEX (IDT)
- 2× ddPCR SuperMix for probes (no dUTP)
- Primers and probes at optimized concentrations
- Semi-skirted 96-well plates (Eppendorf)
- Pierceable foil heat seals
Protocol:
- Reaction Setup: Prepare 22 μL reactions containing 11 μL of 2× ddPCR SuperMix, template DNA (typically 5-10 ng cfDNA equivalent), and optimized primer/probe concentrations determined during assay validation [16].
- Droplet Generation: Generate droplets using the AutoDG instrument according to manufacturer's specifications.
- PCR Amplification: Seal plate and perform PCR using the following typical cycling conditions: 95°C for 10 minutes (enzyme activation); 40 cycles of 94°C for 30 seconds (denaturation) and annealing temperature (specific to assay) for 60 seconds (extension/annealing); 98°C for 10 minutes (enzyme deactivation) [16].
- Post-PCR Processing: Incubate plate at 12°C for minimum 4 hours, then at room temperature for 10 minutes before reading on QX200 droplet reader.
- Quality Controls: Include negative template controls (NTCs: water, TE buffer, elution buffer) and positive template controls (PTCs: wild-type DNA, reference standards) in each run [16].
Data Analysis and Interpretation
Mutation Detection: Analyze using QuantaSoft software. Determine mutant and wild-type droplet populations based on fluorescence amplitude. Calculate mutant copies/μL and variant allele frequency (VAF) using Poisson statistics [16].
Methylation Analysis: For methylation assays, calculate the ratio of methylated to total (methylated + unmethylated) DNA molecules. Apply limit of detection (LOD) thresholds established during validation (typically 0.1% for methylated alleles) [21] [7].
CNV Detection: For copy number analysis, normalize target gene signals to reference genes (e.g., RPP30). Calculate copy number ratios relative to diploid controls, with significant deviations indicating gains or losses [16].
Workflow Visualization
Multiplex ddPCR Biomarker Detection Workflow
Multiplex Assay Development Process
Technical Considerations and Optimization
Assay Design: For point mutation detection, incorporate locked nucleic acid (LNA) bases into probes to enhance discrimination between wild-type and mutant alleles [16]. For methylation assays, design primers to target converted DNA after bisulfite treatment, specifically amplifying methylated sequences [21] [7].
Multiplexing Optimization: When combining assays, systematically optimize primer and probe concentrations to balance amplification efficiency across targets. Address potential oligonucleotide cross-dimerization through in silico analysis and empirical testing [16]. Use distinct fluorescence channels (FAM, HEX) for different targets with appropriate quenchers (e.g., Iowa Black FQ) [16].
Sensitivity and Specificity: Establish limit of blank (LOB) and limit of detection (LOD) for each assay using serial dilutions of positive controls in wild-type background. Implement unique molecular identifiers (UMIs) in upstream processing to distinguish true low-frequency variants from PCR/sequencing errors in highly sensitive applications [4].
False Positive Mitigation: Conduct rigorous negative control experiments to characterize and minimize false positive signals. This is particularly critical for mutation detection at very low VAF (<0.1%) [16]. Pre-PCR processing in dedicated clean rooms and careful reagent preparation can reduce contamination risks.
Multiplex ddPCR represents a robust and reproducible platform for simultaneous detection of point mutations, DNA methylation patterns, and copy number variations in ctDNA. The methodologies outlined herein provide researchers with optimized protocols for implementing this powerful approach in cancer research and drug development. The integration of these complementary biomarker types enables comprehensive molecular profiling from liquid biopsies, supporting applications in treatment response monitoring, resistance mechanism elucidation, and minimal residual disease detection. As the field advances, continued refinement of multiplex ddPCR assays will further enhance their utility in precision oncology workflows.
Circulating tumor DNA (ctDNA) analysis using droplet digital PCR (ddPCR) has emerged as a powerful tool in liquid biopsy, enabling non-invasive cancer monitoring, treatment response assessment, and minimal residual disease detection. This technical note details a standardized workflow for multiplex ddPCR-based ctDNA analysis, framed within broader thesis research on advancing liquid biopsy methodologies. The protocol synthesizes optimized procedures from recent studies to ensure sensitive, reproducible detection of tumor-specific mutations and methylation markers across various cancer types, including lung, colorectal, and pancreatic malignancies. By providing a comprehensive framework from blood collection to data interpretation, this document serves researchers, scientists, and drug development professionals seeking to implement robust ctDNA analysis in both basic and translational research settings.
Pre-Analytical Phase: Blood Collection and Plasma Processing
Blood Collection and Handling
Proper blood collection and processing are critical for preserving ctDNA integrity and preventing genomic DNA contamination. The following standardized protocol ensures optimal sample quality:
Collection Tubes: Collect venous blood using 3-10 mL Streck Cell-Free DNA BCT tubes or standard K₂EDTA vacuum tubes [23] [24]. Streck tubes stabilize nucleated blood cells and prevent lysis, preserving the plasma cfDNA profile for up to 14 days at room temperature.
Processing Timeline: Process samples within 4 hours of venepuncture to minimize background wild-type DNA release from blood cell lysis [6]. Centrifuge at 800-2,000 × g for 10 minutes at room temperature to separate plasma from cellular components [24] [6].
Plasma Isolation: Transfer the supernatant plasma to a fresh tube without disturbing the buffy coat, then perform a second centrifugation at 10,000-16,000 × g for 10 minutes to remove residual cells and debris [6]. Aliquot cleared plasma (typically 1-4 mL) to avoid freeze-thaw cycles and store at -80°C until DNA extraction.
Table 1: Blood Collection and Processing Parameters
| Parameter | Specification | Rationale |
|---|---|---|
| Blood Collection Tube | Streck Cell-Free DNA BCT or K₂EDTA | Prevents cell lysis and preserves ctDNA |
| Processing Time | ≤4 hours post-collection | Minimizes background wild-type DNA contamination |
| Initial Centrifugation | 800-2,000 × g for 10 min | Separates plasma from cellular components |
| Secondary Centrifugation | 10,000-16,000 × g for 10 min | Removes residual cells and platelets |
| Plasma Storage | -80°C in aliquots | Prevents freeze-thaw degradation |
Cell-Free DNA Extraction
Efficient cfDNA extraction maximizes recovery of the short, fragmented ctDNA (typically 90-150 bp) while removing PCR inhibitors:
Extraction Method: Use the QIAamp Circulating Nucleic Acid Kit (Qiagen) or similar silica-membrane based technologies according to manufacturer's instructions [25] [24]. These methods efficiently recover short DNA fragments with consistent yield.
Protocol Modifications: For increased yield, consider modifying standard protocols by increasing plasma input volume (up to 4 mL) and eluting in a smaller volume (50-60 µL) to concentrate cfDNA [25].
Quality Assessment: Quantify cfDNA using fluorometric methods (Qubit dsDNA HS Assay) and assess fragment size distribution. Include exogenous spike-in DNA fragments (e.g., CPP1) at approximately 9,000 copies/mL to monitor extraction efficiency [6]. Assess potential genomic DNA contamination using an immunoglobulin gene-specific ddPCR assay [6].
Table 2: cfDNA Extraction and Quality Control
| Component | Specification | Purpose |
|---|---|---|
| Extraction Kit | QIAamp Circulating Nucleic Acid Kit | Optimized for short fragment recovery |
| Plasma Input | 2-4 mL | Maximizes ctDNA yield |
| Elution Volume | 50-60 µL | Concentrates cfDNA for analysis |
| Spike-in Control | CPP1 DNA fragment (~9,000 copies/mL) | Monitors extraction efficiency |
| gDNA Contamination Check | Immunoglobulin gene ddPCR assay | Detects white blood cell contamination |
Analytical Phase: Assay Design and ddPCR Setup
Mutation Detection Assays
Two primary approaches are employed for ctDNA detection:
Tumor-Informed Assays: Design patient-specific ddPCR assays based on mutations identified in tumor tissue sequencing (e.g., TP53, KRAS, BRAF, NRAS, EGFR) [24] [26]. This approach enables highly sensitive monitoring of known tumor-specific variants.
Tumor-Agnostic Methylation Panels: Develop multiplex ddPCR assays targeting cancer-specific methylation patterns (e.g., HOXA9 and other hypermethylated regions) identified through bioinformatic analysis of public methylation arrays [6]. This method is particularly valuable when tumor tissue is unavailable.
Multiplex ddPCR Assay Configuration
The ddPCR workflow involves partitioning samples into thousands of nanodroplets, enabling absolute quantification of target molecules:
Reaction Setup: Prepare 22-40 µL reactions containing 11 µL of 2× ddPCR Supermix for Probes, 1-2 µL of primer-probe mix (final concentration 250-900 nM primers, 100-250 nM probes), and 2-9 µL of template cfDNA (approximately 5-50 ng) [6] [24]. Include no-template controls (NTC) and wild-type-only controls in each run.
Droplet Generation: Generate 20,000 droplets per sample using the QX200 Droplet Generator. Emulsified reactions undergo thermal cycling with optimized conditions: 95°C for 10 minutes (1 cycle); 94°C for 30 seconds and assay-specific annealing temperature (55-60°C) for 60 seconds (40-55 cycles); 98°C for 10 minutes (1 cycle); and final hold at 12°C [24] [6].
Droplet Reading and Analysis: Read plates using the QX200 Droplet Reader and analyze with QuantaSoft software (v1.7.4 or higher). Set fluorescence amplitude thresholds based on positive and negative control clusters to distinguish mutant and wild-type populations.
Post-Analytical Phase: Data Analysis and Interpretation
Quality Control Criteria
Implement stringent quality control measures to ensure data reliability:
- Droplet Count: Accept wells with >10,000 total droplets; repeat analyses with fewer droplets [24].
- False-Positive Rate: Establish mutation-specific thresholds using wild-type-only control samples (typically 0-1 false-positive droplets per assay) [24].
- Extraction Efficiency: Verify using spike-in controls; >70% recovery is acceptable.
- Background Estimation: Calculate limit of detection (LOD) and limit of blank (LOB) for each assay using negative controls.
Quantification and Statistical Analysis
- Absolute Quantification: Calculate mutant and wild-type copies/μL using Poisson distribution statistics applied to positive and negative droplet counts [24].
- Variant Allele Frequency (VAF): Determine using the formula: VAF = [mutant copies/(mutant copies + wild-type copies)] × 100%.
- Clinical Cut-offs: Establish assay-specific thresholds for positivity; studies commonly use VAF ≥0.01% to 0.1% for mutation detection and ≥1% for prognostic stratification [26] [23].
Table 3: Key Performance Metrics for ctDNA Detection by ddPCR
| Performance Metric | Typical Range | Clinical/Research Utility |
|---|---|---|
| Limit of Detection (LOD) | 0.01%-0.1% VAF | Enables MRD and early-stage cancer detection |
| Absolute Sensitivity | 2-422 mutant copies/mL plasma | Varies by tumor burden and cancer type |
| Detection Rate in Advanced Cancer | 54%-96% | Depends on cancer type and assay design |
| Detection Rate in Early-Stage Cancer | 38.7%-46.8% | Lower tumor shedding affects sensitivity |
| Assay Turnaround Time | 7-20 hours | Faster than NGS (days to weeks) |
Research Reagent Solutions
Table 4: Essential Research Reagents for ctDNA ddPCR Analysis
| Reagent/Category | Specific Examples | Function in Workflow |
|---|---|---|
| Blood Collection Tubes | Streck Cell-Free DNA BCT, K₂EDTA tubes | Stabilizes blood cells and preserves ctDNA |
| cfDNA Extraction Kits | QIAamp Circulating Nucleic Acid Kit (Qiagen), DSP Circulating DNA Kit | Isolves and purifies fragmented cfDNA |
| ddPCR Master Mixes | ddPCR Supermix for Probes (no dUTP) (Bio-Rad) | Provides enzymes and reagents for PCR |
| Bisulfite Conversion Kits | EZ DNA Methylation-Lightning Kit (Zymo Research) | Converts unmethylated cytosines for methylation assays |
| Target-Specific Reagents | Custom primer-probe sets, ddPCR Mutation Assays | Detects specific mutations or methylation markers |
| Quality Control Assays | Exogenous spike-in controls (CPP1), gDNA contamination assays | Monitors extraction efficiency and sample quality |
Applications in Cancer Research
The optimized ddPCR workflow for ctDNA analysis supports multiple research applications:
Treatment Response Monitoring: Serial ctDNA quantification during therapy correlates with tumor burden changes, often preceding radiographic response assessment [4] [26]. Declining ctDNA levels predict favorable outcomes, while persistent detection indicates resistance.
Minimal Residual Disease (MRD) Detection: Post-treatment ctDNA assessment identifies molecular residual disease with higher sensitivity than imaging, stratifying recurrence risk [27] [23]. Patients with ctDNA-positive status after curative-intent surgery have significantly higher recurrence risk.
Therapeutic Target Identification: Multiplex ddPCR panels can simultaneously screen for multiple actionable mutations (e.g., EGFR, KRAS, BRAF) to guide targeted therapy selection [28] [23].
Clonal Evolution Tracking: Longitudinal monitoring detects emerging resistance mutations (e.g., EGFR T790M), enabling timely therapy adjustments [27] [4].
The workflow presented establishes a standardized framework for sensitive ctDNA detection using multiplex ddPCR, facilitating implementation in cancer research and accelerating the development of liquid biopsy applications in precision oncology.
Assay Development and Translational Applications in Oncology Research
The analysis of circulating tumor DNA (ctDNA) has emerged as a cornerstone of liquid biopsy applications in oncology, enabling non-invasive cancer detection, therapy selection, and disease monitoring. Droplet digital PCR (ddPCR) offers an exceptionally sensitive and absolute quantification platform for ctDNA detection, with multiplexing providing enhanced capabilities for simultaneous assessment of multiple biomarkers. This approach is particularly valuable given the low abundance of ctDNA in plasma, especially in early-stage disease or minimal residual disease settings [6] [29]. The development of robust multiplex ddPCR assays requires careful consideration of panel selection based on genomic or epigenomic alterations, meticulous primer and probe design, and rigorous validation strategies to ensure clinical utility. When properly designed, these assays can detect ctDNA with variant allele frequencies as low as 0.003% [29], demonstrating the powerful sensitivity achievable through optimized multiplex approaches.
Panel Selection Strategies
Biomarker Selection Criteria
Selecting appropriate biomarkers forms the foundation of any successful multiplex ddPCR assay. The choice between mutation-based and methylation-based panels depends on the specific clinical application and cancer type.
Mutation-based panels target somatic mutations specific to an individual's cancer, making them ideal for patient-specific monitoring. For example, in the TRICIA trial for triple-negative breast cancer, researchers identified a median of 15 mutations per patient via whole-exome sequencing before selecting a single truncal mutation for ctDNA detection via ddPCR [30]. This patient-informed approach enables highly specific tracking of residual disease.
Methylation-based panels exploit the predictable and recurrent nature of DNA methylation changes in cancer. A key advantage is their potential for "off-the-shelf" use without requiring prior knowledge of a patient's tumor genetics [7]. One study developed a multiplex assay targeting five tumor-specific methylation markers for lung cancer detection, four identified through bioinformatics analysis of Illumina 450K methylation arrays and one (HOXA9) from previous research [6]. Similarly, a multi-cancer detection assay was developed using three differentially methylated regions to detect eight cancer types with a cross-validated area under the curve of 0.948 [7].
Computational Design andIn SilicoAnalysis
Bioinformatics pipelines are crucial for identifying optimal marker combinations. The development process typically begins with in silico analysis of public methylation databases such as The Cancer Genome Atlas (TCGA) to identify differentially methylated CpG sites [6] [7]. One established workflow involves:
- Data Acquisition: Gathering methylation array data (e.g., Infinium HumanMethylation450 BeadChip) from tumor samples, normal adjacent tissue, and normal blood cells [6].
- Differential Analysis: Calculating mean beta-value differences between tumor and normal samples, typically selecting sites with differences >0.5 [6].
- Feature Selection: Applying recursive feature elimination with cross-validation to identify the most discriminatory markers [6].
A critical consideration in multiplex design is the fundamental tradeoff between multiplexing level and coverage. Research has demonstrated a computational phase transition where assay design becomes dramatically more difficult when the probability of primer pair interactions exceeds a critical threshold [31]. This limitation can be mitigated by having a larger pool of candidate markers or loosening primer selection constraints, though the latter may introduce other adverse effects.
Table 1: Performance Characteristics of Published Multiplex ddPCR Assays
| Cancer Type | Assay Type | Markers | Sensitivity | Specificity | Reference |
|---|---|---|---|---|---|
| Lung Cancer | Methylation-specific multiplex | 5 methylation markers | 38.7-46.8% (non-metastatic); 70.2-83.0% (metastatic) | >95% | [6] |
| 8 Cancer Types | Methylation-based multi-cancer | 3 methylation targets | 53.8-100% across cancer types | 80-100% across cancer types | [7] |
| Triple-Negative Breast Cancer | Tumor-informed mutation detection | Patient-specific truncal mutations | 97% detection before clinical relapse | 100% in RCB 3 patients | [30] |
| Early Breast Cancer | Mutation detection with increased blood volume | Patient-specific mutations | 100% pre-treatment detection | N/A | [29] |
Primer and Probe Design
Design Principles and Considerations
The three-phase development process for non-competing multiplex dPCR assays using target-specific fluorescently labeled hydrolysis probes involves specific design considerations to overcome unique challenges associated with multiplexing in dPCR [32]:
Phase 1: In Silico Assay Design
- Target-specific primers and probes are selected or designed, typically using specialized software.
- Potential issues with primer and probe interactions are identified through comprehensive in silico analysis.
- Fluorophores and quenchers are chosen based on the specific dPCR instrumentation available, ensuring spectral compatibility.
- Careful attention is paid to avoiding primer-dimer formations and other non-specific interactions that can compromise assay performance.
Phase 2: Wet-lab Validation
- Assays are benchmarked using positive controls with known characteristics.
- Insufficient performance leads to iterative assay redesign as needed.
- Optimal annealing temperatures and reaction conditions are established empirically.
Phase 3: Assay Implementation
- Assay specificity and sensitivity are validated on relevant sample matrices.
- Limit of detection (LOD) and limit of blank (LOB) are established [7].
A critical consideration is that achieving broad SNP coverage rapidly transitions from "very easy to very hard" as the target multiplexing level increases [31]. The presence of this computational phase transition suggests fundamental limits to scaling multiplex PCR performance for high-throughput applications.
Fluorophore Selection and Multiplexing Capacity
The choice of fluorophores is constrained by the available channels on ddPCR systems. Assays typically utilize fluorescein (FAM)-based and hexachlorofluorescein (HEX)-based probes, with newer systems offering additional channels. A multi-cancer detection approach successfully implemented a combination of triplex and duplex ddPCR assays, with output data from both assays combined to obtain a comprehensive read-out from three targets together [7]. This strategy effectively increases the multiplexing capacity beyond the limitations of a single reaction.
Wet-Lab Validation Strategies
Analytical Validation
Comprehensive validation is essential to establish assay performance characteristics. Key parameters include:
Sensitivity and Specificity Determination: Using samples with known mutation status to establish true positive and true negative rates. For example, one methylation-based multiplex assay demonstrated 100% sensitivity and 100% specificity in Residual Cancer Burden 3 triple-negative breast cancer patients [30].
Limit of Detection (LOD) and Limit of Blank (LOB) Establishment: Through dilution series of positive control material in wild-type background. In one study, the minimum variant allele frequency for ctDNA detection was 0.01% in pre-treatment early breast cancer samples [29].
Precision and Reproducibility Assessment: Measuring intra-assay and inter-assay coefficients of variation (%CV) [7].
Dynamic Range Evaluation: Verifying accurate quantification across clinically relevant concentrations.
Table 2: Essential Validation Parameters for Multiplex ddPCR Assays
| Validation Parameter | Experimental Approach | Acceptance Criteria |
|---|---|---|
| Analytical Sensitivity | Dilution series of positive control material | LOD of 0.01% VAF or lower |
| Analytical Specificity | Testing against known negative samples | >98% specificity |
| Precision | Replicate testing of samples across multiple runs | <10% CV for copy number quantification |
| Dynamic Range | Samples with varying mutant allele frequencies | Linear response across 4 orders of magnitude |
| Robustness | Variation in reaction conditions (e.g., annealing temperature) | Consistent performance with ±2°C variation |
Clinical Validation
Clinical validation establishes assay performance in real-world scenarios. The COMBI-AD trial for stage III melanoma validated ddPCR assays for detecting BRAFV600-mutant ctDNA, finding that baseline ctDNA detection was associated with significantly worse recurrence-free survival (HR 2.91-2.98) and overall survival (HR 3.35-4.27) [33]. Similarly, in the TRICIA trial for triple-negative breast cancer, lack of ctDNA detection post-neoadjuvant chemotherapy pre-operatively was highly prognostic, with 95% distant-disease relapse-free survival [30].
Longitudinal monitoring represents another crucial validation aspect, with studies demonstrating that ctDNA dynamics can predict treatment response and anticipate clinical relapse by several months [29] [33]. One study found that ddPCR measurements of ctDNA during follow-up could identify patients at high risk of early recurrence, with patients having adverse longitudinal ctDNA kinetics showing markedly shorter median recurrence-free survival (5.32-8.31 months) compared to those with favorable kinetics (19.25 months to not reached) [33].
Experimental Protocols
Sample Processing and DNA Extraction
Plasma Collection and Processing:
- Collect whole blood in EDTA or specialized cell-free DNA blood collection tubes.
- Process samples within 4 hours of venepuncture by centrifugation at 2,000 × g for 10 minutes to separate plasma [6].
- Aliquot plasma and store at -80°C until extraction.
- For increased sensitivity, consider larger plasma volumes (20-40 mL instead of conventional 5-10 mL) [29].
Cell-free DNA Extraction:
- Thaw plasma at 5°C and centrifuge at 10,000 × g for 10 minutes to remove debris.
- Add exogenous spike-in DNA (e.g., ~9000 copies/mL of CPP1) to monitor extraction efficiency [6].
- Extract cfDNA using commercial kits (e.g., DSP Circulating DNA Kit on QIAsymphony SP) according to manufacturer's instructions [6].
- Elute DNA in 60 μL elution buffer.
- Concentrate extracted DNA to 20 μL using centrifugal filter units [6].
Bisulfite Conversion (for Methylation Assays)
- Use commercial bisulfite conversion kits (e.g., EZ DNA Methylation-Lightning Kit) [6] [7].
- Convert 20 ng DNA per sample according to manufacturer's instructions [7].
- Elute bisulfite-converted DNA in 15-25 μL M-Elution Buffer [6] [7].
- Use converted DNA within 10 days, storing at -20°C [7].
Droplet Digital PCR
Reaction Setup:
- Prepare ddPCR reaction mix containing ddPCR Supermix, primers, and probes.
- Typically use 20 ng bisulfite-converted DNA or equivalent volume of eluted cfDNA per reaction.
- Partition samples into droplets using automated droplet generators.
Thermal Cycling:
- Program thermal cycler with appropriate protocol for bisulfite-converted DNA (if applicable) or standard ddPCR conditions.
- Common conditions: 95°C for 10 minutes (enzyme activation), followed by 40 cycles of 94°C for 30 seconds (denaturation) and appropriate annealing temperature (55-60°C) for 1 minute (annealing/extension), with a final 98°C for 10 minutes (enzyme deactivation) [6] [7].
- Ramp rate of 2°C/second is typically used.
Droplet Reading and Analysis:
- Read plates using droplet readers.
- Analyze data with companion software using appropriate threshold settings.
- For multiplex assays, use two different cut-off methods to determine ctDNA status and examine their effects on sensitivity and specificity [6].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for Multiplex ddPCR Assays
| Reagent/Material | Function | Examples/Specifications |
|---|---|---|
| Nucleic Acid Extraction Kits | Isolation of high-quality cfDNA from plasma | DSP Circulating DNA Kit (Qiagen), QIAamp DNA Micro Kit (Qiagen) [6] [7] |
| Bisulfite Conversion Kits | Conversion of unmethylated cytosines to uracils for methylation analysis | EZ DNA Methylation-Lightning Kit (Zymo Research) [6] [7] |
| ddPCR Supermix | Provides optimal reaction environment for digital PCR | ddPCR Supermix for Probes (Bio-Rad) |
| Hydrolysis Probes | Sequence-specific detection with fluorescent reporters | FAM, HEX, or other dye-labeled TaqMan-style probes [32] |
| Primer Sets | Target-specific amplification | HPLC-purified primers designed for bisulfite-converted sequences (if applicable) |
| Droplet Generation Oil | Creates water-in-oil emulsions for partitioning | Droplet Generation Oil for Probes (Bio-Rad) |
| Positive Control Materials | Assay validation and quality control | Synthetic oligonucleotides, cell line DNA (HCT116, Cal27) [7] |
| Exogenous Spike-in DNA | Monitoring extraction efficiency and potential inhibition | CPP1 spike-in fragment (~9000 copies/mL) [6] |
Well-designed multiplex ddPCR assays represent powerful tools for ctDNA analysis in cancer research and clinical applications. Successful implementation requires integrated consideration of computational design, wet-lab optimization, and rigorous validation. The strategies outlined herein provide a framework for developing robust assays capable of detecting rare ctDNA molecules with high specificity. As the field advances, further refinement of multiplexing approaches will continue to enhance our ability to monitor cancer dynamics through liquid biopsies, ultimately supporting more personalized treatment approaches. Future directions include increasing multiplexing capacity through novel chemistries and fluorophores, standardizing protocols across platforms, and demonstrating clinical utility in prospective interventional trials.
The analysis of circulating tumor DNA (ctDNA) from liquid biopsies represents a transformative approach in oncology, enabling non-invasive cancer detection, prognosis, and monitoring of treatment response [6] [7]. Cell-free DNA (cfDNA) fragments released into the bloodstream carry tumor-specific epigenetic signatures, with aberrant DNA methylation being one of the most promising biomarker classes due to its early occurrence in carcinogenesis and high recurrence across cancer types [6] [34] [7].
This application note details a standardized pipeline for preparing bisulfite-converted cfDNA, a critical precursor for downstream methylation-specific droplet digital PCR (ddPCR) analysis. The optimized protocols and quality control measures outlined herein ensure the generation of high-quality, analysis-ready DNA, supporting the accuracy and reliability of multiplex ddPCR assays in ctDNA research and drug development.
The complete sample processing pipeline, from blood collection to analysis-ready bisulfite-converted DNA, involves several integrated stages. The following diagram illustrates this workflow and the critical logical relationships between each step:
Detailed Experimental Protocols
Blood Collection and Plasma Separation
Principle: Stabilize blood samples and isolate plasma to prevent genomic DNA contamination from leukocytes, preserving the native fragment profile of cfDNA.
Materials:
- K₂EDTA or Streck Cell-Free DNA BCT blood collection tubes
- Refrigerated centrifuge
- Sterile pipettes and nuclease-free microtubes
Procedure:
- Collection: Draw venous blood into K₂EDTA tubes. Invert gently 8-10 times to mix.
- Initial Spin: Centrifuge at 2,000 × g for 10 minutes at 4°C within 4 hours of venipuncture [6].
- Plasma Transfer: Carefully transfer the upper plasma layer to a nuclease-free tube using a sterile pipette, avoiding the buffy coat.
- Second Spin: Centrifuge the transferred plasma at 10,000 × g for 10 minutes to remove any residual cells [6].
- Storage: Aliquot cleared plasma and store at -80°C if not processed immediately.
cfDNA Extraction
Principle: Efficiently isolate short-fragment cfDNA from large-volume plasma samples while excluding high molecular weight genomic DNA.
Materials:
- Recommended Kits: QIAamp Circulating Nucleic Acid Kit (CNA kit) or Maxwell RSC ccfDNA Plasma Kit [34]
- Magnetic rack for 24-well plates (if using magnetic bead-based methods)
- Heated shaker
- Nuclease-free elution buffer (e.g., TE buffer, AVE buffer)
Procedure (Manual, CNA Kit):
- Prepare Sample: Mix 1-5 mL plasma with Proteinase K and buffer ACL.
- Incubate: Incubate at 60°C for 30 min.
- Bind DNA: Add ethanol, then load mixture onto QIAamp Mini column.
- Wash: Centrifuge and wash with buffers AW1 and AW2.
- Elute: Add elution buffer to the column membrane, incubate, and centrifuge to recover cfDNA.
Procedure (Automated, High-Throughput):
- Platform Setup: Configure a liquid handling robot (e.g., Tecan Freedom EVO 200) with alternating 5 mL and 1 mL dilutors and a centric gripper [35].
- DNA Binding: Perform magnetic bead-based DNA extraction in 24-well plates on flat heated shakers [35].
- Bead Capture: Use a 24-well magnet plate to capture beads, then transfer to a 96-well deep well plate for volume reduction [35].
- Elution: Elute DNA in a 96-well PCR plate for seamless integration with downstream steps [35].
Pre-Bisulfite Conversion Quality Control
Principle: Quantify and qualify extracted cfDNA to ensure it meets the requirements for successful bisulfite conversion and downstream ddPCR.
Materials:
- Fluorometer (e.g., Qubit Fluorometer 2.0) with dsDNA HS Assay Kit
- Bioanalyzer system (e.g., Agilent 2100) with High Sensitivity DNA kit
- Droplet digital PCR system
Procedure:
- Quantification: Use Qubit fluorometer for accurate concentration measurement.
- Fragment Analysis: Run 1 µL cfDNA on Bioanalyzer to confirm peak size ~166 bp (characteristic of cfDNA) [34].
- gDNA Contamination Check: Utilize ddPCR assays amplifying different amplicon lengths (e.g., 65 bp vs. 250 bp region of EMC7 gene); a high ratio of long to short fragments indicates gDNA contamination [6].
- Extraction Efficiency (Optional): Add exogenous spike-in DNA (e.g., CPP1) before extraction and quantify recovery with a specific ddPCR assay [6].
Bisulfite Conversion and Purification
Principle: Treat DNA with bisulfite to deaminate unmethylated cytosines to uracils, while methylated cytosines remain unchanged, enabling methylation status determination by subsequent PCR.
Materials:
- Recommended Kits: EpiTect Plus DNA Bisulfite Kit or EZ DNA Methylation-Lightning Kit [6] [34] [7]
- Thermal cycler
- Nuclease-free water
Procedure (EpiTect Plus DNA Bisulfite Kit):
- Denature DNA: Mix up to 2 µg DNA with buffer BD and incubate at 95°C for 5 min.
- Prepare Bisulfite Mix: Combine bisulfite solution and DNA protection buffer.
- Convert: Add bisulfite mix to denatured DNA, incubate in thermal cycler (cycling conditions: 95°C for 5 min, 60°C for 25 min, 95°C for 5 min, 60°C for 85 min, 95°C for 5 min, 60°C for 175 min, hold at 20°C).
- Bind DNA: Transfer reaction to a spin column containing buffer BL.
- Desulphonate: Wash with buffer BW, then incubate with buffer BD for 20 min at room temperature.
- Wash and Elute: Wash with buffer BW, then elute with buffer EB.
Procedure (Automated High-Throughput):
- Automation: Adapt manual bisulfite conversion steps for liquid handling robots.
- Interlaced Processing: Implement a highly interlaced 4 × 24 sample protocol for DNA extraction, bisulfite conversion, and purification in a 96-well plate format [35].
- Walk-Away Solution: Process 96 samples in approximately 7 hours 30 minutes with minimal manual intervention [35].
Post-Conversion Quality Control
Principle: Assess the quantity, conversion efficiency, and PCR-amplifiability of the final bisulfite-converted DNA (bisDNA).
Materials:
- Droplet digital PCR system
- Control assays (e.g., 4Plex control assay, MYOD1 control assay, β-actin assay for bisulfite-converted sequence)
- Electrophoresis system (e.g., Bioanalyzer)
Procedure:
- Quantification: Use ddPCR with control assays (e.g., 4Plex) to accurately quantify the amount of recovered bisDNA [34].
- Conversion Efficiency: Perform ddPCR with methylation-specific assays for control DNA with known methylation status.
- PCR-Amplifiability: Test bisDNA with a ddPCR assay targeting a bisulfite-converted sequence of a reference gene (e.g., β-actin) [35].
- Fragment Length Analysis (Optional): Assess fragment size distribution using Bioanalyzer to confirm absence of excessive degradation.
Performance Data and Kit Comparison
Comparative Performance of Bisulfite Conversion Kits
The efficiency of bisulfite conversion directly impacts DNA recovery and downstream assay performance. The following table summarizes the quantitative performance of leading commercial kits:
Table 1: Performance Comparison of Bisulfite Conversion Kits [34]
| Bisulfite Conversion Kit | Average DNA Recovery (20 ng input) | Relative DNA Yield | Key Characteristics |
|---|---|---|---|
| EpiTect Plus DNA Bisulfite Kit | 10-20% | Highest | Highest yield and recovery across input amounts |
| Premium Bisulfite Kit | 10-20% | High | Good performance, especially at lower inputs (2-0.5 ng) |
| EZ DNA Methylation-Direct Kit | <10-20% | High | Good performance at higher inputs (20-3 ng) |
| EpiJET Bisulfite Conversion Kit | <10% | Low | Lower yield across all input amounts |
| Imprint DNA Modification Kit | <10% | Lowest | Lowest recovery |
Comparative Performance of cfDNA Isolation Kits
The choice of isolation method significantly affects cfDNA yield and purity. The following table compares the performance of three commercially available kits:
Table 2: Performance Comparison of cfDNA Isolation Kits [34]
| cfDNA Isolation Kit | Total cfDNA Yield (from 1 mL plasma) | Average Fragment Size (bp) | Level of gDNA Contamination |
|---|---|---|---|
| QIAamp Circulating Nucleic Acid Kit (CNA) | ~13.9 ng (plasma only) | 165-170 | Higher |
| QIAamp MinElute ccfDNA Mini Kit | ~5.0 ng (plasma only) | 174-177 | Lower |
| Maxwell RSC ccfDNA Plasma Kit | ~5.2 ng (plasma only) | 174-177 | Lower |
Optimal Kit Combinations for Methylation Analysis
Based on systematic evaluation, the combination of the QIAamp Circulating Nucleic Acid Kit (CNA) for cfDNA isolation and the EpiTect Plus DNA Bisulfite Kit for conversion was identified as the best-performing workflow, yielding the highest amount of bisulfite-converted cfDNA suitable for downstream methylation-specific ddPCR [34].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for cfDNA Methylation Analysis
| Item | Function/Application | Example Products/Assays |
|---|---|---|
| cfDNA Isolation Kits | Isolation of cell-free DNA from plasma/serum | QIAamp Circulating Nucleic Acid Kit (CNA), Maxwell RSC ccfDNA Plasma Kit [34] |
| Bisulfite Conversion Kits | Conversion of unmethylated cytosine to uracil | EpiTect Plus DNA Bisulfite Kit, EZ DNA Methylation-Lightning Kit [6] [34] |
| Methylation-Specific ddPCR Assays | Detection and absolute quantification of methylated alleles | Custom assays for targets like HOXA9, BCAT1, IKZF1 [6] [34] [7] |
| Quality Control Assays | Assessing DNA quantity, fragmentation, and conversion efficiency | EMC7 assays (65 bp/250 bp), 4Plex control assay, β-actin bisulfite-converted assay [35] [6] [34] |
| Automated Liquid Handling | High-throughput, reproducible sample processing | Tecan Freedom EVO 200 platform with customized shakers and magnet plates [35] |
Troubleshooting Guide
Table 4: Common Issues and Recommended Solutions
| Problem | Potential Cause | Solution |
|---|---|---|
| Low cfDNA yield after extraction | Insfficient plasma volume, inefficient binding | Increase plasma input volume (e.g., 3-5 mL); ensure correct ethanol concentration during binding [35] [34] |
| High gDNA contamination | Incomplete plasma separation; lysis of white blood cells | Perform double-centrifugation of plasma; avoid disturbing buffy coat; use kits designed for cfDNA [6] |
| Low DNA recovery after bisulfite conversion | Inefficient purification; DNA degradation | Use high-performance kits (e.g., EpiTect Plus); ensure fresh bisulfite reagent; avoid over-drying columns [34] |
| Poor amplification in ddPCR | Incomplete bisulfite conversion; inhibitor carryover | Include control for conversion efficiency; perform additional purification steps; ensure proper elution buffer pH [35] |
| High variability between replicates | Pipetting errors; inconsistent bead handling | Automate process using liquid handling robots; standardize incubation and mixing times [35] |
The early detection of cancer is a critical factor in improving patient survival outcomes. Currently, routine screening is recommended for only a few cancer types, leaving approximately 70% of cancer diagnoses and deaths associated with cancers without established screening protocols [36] [37]. Multi-cancer early detection (MCED) tests represent a transformative approach to cancer screening by enabling the simultaneous detection of multiple cancer types through minimally invasive liquid biopsy. These tests analyze circulating tumor DNA (ctDNA) released into the bloodstream by tumor cells, leveraging tumor-specific genetic and epigenetic alterations as biomarkers [38] [4].
Droplet digital PCR (ddPCR) has emerged as a particularly powerful technology for ctDNA analysis due to its high sensitivity, absolute quantification capabilities, and robustness in detecting rare targets in background wild-type DNA [9]. When applied to MCED, multiplex ddPCR assays can screen for multiple cancer-specific markers simultaneously, offering a promising tool for population-scale cancer screening. Furthermore, the patterns of detected biomarkers can provide clues about the tissue of origin (TOO), guiding subsequent diagnostic workups [39]. This application note details experimental protocols and performance data for implementing multiplex ddPCR in MCED and TOO detection workflows, providing researchers with practical frameworks for advancing ctDNA-based cancer detection research.
Performance Characteristics of MCED Tests
The clinical validity of MCED approaches is demonstrated through their sensitivity and specificity in detecting multiple cancer types across stages. The tables below summarize key performance metrics from recent studies.
Table 1: Performance of DNA Methylation-Based MCED ddPCR Test for Four Cancers [39]
| Cancer Type | Sensitivity (All Stages) | Sensitivity (Early Stage) | Specificity |
|---|---|---|---|
| Lung | 81.82% | Slightly lower (specific value not reported) | 91.04% |
| Colorectal | 69.23% | Slightly lower (specific value not reported) | 91.04% |
| Breast | 45.00% | Slightly lower (specific value not reported) | 91.04% |
| Prostate | 44.14% | Slightly lower (specific value not reported) | 91.04% |
| Overall Panel | 60.10% | - | 87.40% |
Table 2: Projected Impact of Widespread MCED Testing on Cancer Staging [36]
| Cancer Stage | Projected Change in Diagnosis with MCED Testing |
|---|---|
| Stage I | 10% increase |
| Stage II | 20% increase |
| Stage III | 34% increase |
| Stage IV | 45% decrease |
Table 3: Performance of a Commercial Multi-Analyte MCED Test [37]
| Performance Metric | Result |
|---|---|
| Overall Sensitivity | 64.1% |
| Sensitivity for Six High-Risk Cancers* | 67.8% |
| Specificity | 97.4% |
| Turnaround Time | ~2 weeks |
*Pancreatic, esophageal, liver, lung, stomach, and ovarian cancers.
Experimental Protocols
Protocol 1: Multiplex ddPCR for MCED Using DNA Methylation Markers
This protocol is adapted from studies detecting multiple cancer types using hypermethylated gene promoters [39] [6].
Sample Collection and Processing
- Blood Collection: Draw whole blood (10-20 mL recommended) into EDTA-containing tubes to prevent coagulation.
- Plasma Separation: Centrifuge tubes at 1,600 × g for 15 minutes at 4°C within 4 hours of collection to separate plasma from cellular components.
- Aliquoting and Storage: Transfer supernatant (plasma) to sterile tubes without disturbing the buffy coat. Aliquot to avoid freeze-thaw cycles and store at -80°C until cfDNA extraction.
Cell-free DNA Extraction
- Thaw Plasma: Thaw plasma aliquots at 5°C and centrifuge at 10,000 × g for 10 minutes to remove residual cells and debris [6].
- cfDNA Extraction: Extract cfDNA from 4 mL plasma using commercially available kits (e.g., DSP Circulating DNA Kit on QIAsymphony SP) according to manufacturer's instructions.
- Elution: Elute cfDNA in 50-60 μL of elution buffer. Concentrate to 20 μL using Amicon Ultra-0.5 Centrifugal Filter Units if necessary [6].
- Quantification and Quality Control: Quantify cfDNA concentration using a fluorescence-based assay. Assess fragment size distribution (expected peak ~166 bp) via bioanalyzer.
Bisulfite Conversion
- Conversion Reaction: Treat extracted cfDNA with bisulfite using the EZ DNA Methylation-Lightning Kit to convert unmethylated cytosines to uracils, while methylated cytosines remain unchanged.
- Purification: Purify bisulfite-converted DNA according to kit instructions.
- Elution: Elute in 15 μL M-Elution Buffer [6].
Multiplex ddPCR Assay
- Reaction Setup: Prepare a 15-25 μL reaction mixture containing:
- 5 μL bisulfite-converted DNA template
- 1× ddPCR Supermix for Probes
- Primer and probe mixes for target methylation markers (e.g., ADCY4, MIR129-2, NID2, MAGI2 for lung, breast, colorectal, and prostate cancers) [39]
- Nuclease-free water to volume
- Droplet Generation: Transfer reaction mixture to a DG32 cartridge and generate approximately 20,000 nanodroplets using a droplet generator.
- PCR Amplification: Perform amplification on a thermal cycler with the following conditions:
- 95°C for 5 minutes (enzyme activation)
- 40 cycles of:
- 95°C for 15 seconds (denaturation)
- 60°C for 30-60 seconds (annealing/extension)
- 98°C for 10 minutes (enzyme deactivation)
- 4°C hold
- Droplet Reading: Transfer stabilized droplets to a droplet reader for fluorescence detection in multiple channels (FAM, HEX, etc.).
Data Analysis
- Threshold Setting: Analyze fluorescence amplitude data using manufacturer's software. Set positivity thresholds based on negative controls and no-template controls.
- Absolute Quantification: Calculate copies/μL of methylated targets based on Poisson distribution analysis of positive and negative droplets.
- Result Interpretation: Apply predetermined cut-off values for calling samples positive for cancer-associated methylation.
Diagram 1: MCED ddPCR Workflow (10 steps)
Protocol 2: Universal Sarcoma Detection via Methylated ctDNA
This protocol describes a specialized approach for detecting sarcomas using universally methylated ctDNA markers [40].
Sample Processing
- Follow the same sample collection and cfDNA extraction procedures as in Protocol 3.1.1 and 3.1.2.
Bisulfite Conversion and ddPCR
- Bisulfite Treatment: Perform bisulfite conversion as in Protocol 3.1.3.
- Multiplex Assay Setup: Prepare ddPCR reaction targeting seven hypermethylated genomic positions specific to sarcomas.
- Droplet Generation and Amplification: Follow steps in Protocol 3.1.4.
Analytical Validation
- Sensitivity Assessment: Validate assay sensitivity through serial dilutions (demonstrated detection at 1:1,000 dilution with methylated allele frequency of 0.06%) [40].
- Specificity Confirmation: Verify minimal background in non-sarcoma tissues and white blood cells.
- Dynamic Monitoring: Apply assay to longitudinal samples for treatment response monitoring.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents and Materials for MCED ddPCR Assays
| Reagent/Material | Function | Examples/Specifications |
|---|---|---|
| EDTA Blood Collection Tubes | Prevents coagulation and preserves cell-free DNA | 10-20 mL draw volume |
| cfDNA Extraction Kit | Isolation of high-quality cell-free DNA from plasma | DSP Circulating DNA Kit (Qiagen) |
| Bisulfite Conversion Kit | Converts unmethylated cytosine to uracil for methylation analysis | EZ DNA Methylation-Lightning Kit |
| ddPCR Supermix | Provides optimal environment for PCR in droplets | ddPCR Supermix for Probes (no dUTP) |
| Methylation-Specific Primers/Probes | Detects cancer-specific hypermethylated regions | ADCY4, MIR129-2, NID2, MAGI2 assays |
| Droplet Generation Oil | Creates stable water-in-oil emulsions for partitioning | DG32 Droplet Generation Oil |
| ddPCR Plates/Cartridges | Holds reaction mixture for droplet generation | DG32 Cartridges |
| Positive Control DNA | Validates assay performance | Synthetic methylated DNA fragments |
Tissue of Origin Determination
Accurate tissue of origin determination is crucial for guiding diagnostic follow-up after a positive MCED result. The approach relies on detecting cancer-specific methylation patterns that are characteristic of different tumor types [39] [6].
Diagram 2: Tissue of Origin Determination (5 steps)
The process involves:
- Pattern Recognition: Analyzing the combination and relative abundance of different methylation markers detected in the positive sample.
- Algorithmic Classification: Using trained classifiers to match the detected methylation profile to known cancer type signatures.
- Diagnostic Guidance: Directing subsequent imaging and diagnostic procedures to the most likely tissue of origin, improving diagnostic efficiency.
Multiplex ddPCR represents a robust, sensitive, and clinically actionable technology for multi-cancer early detection. The protocols outlined herein provide researchers with validated methodologies for implementing MCED assays targeting DNA methylation biomarkers. The high specificity and moderate to high sensitivity across multiple cancer types, particularly for lethal malignancies with no current screening options, position this technology as a promising complement to existing cancer screening paradigms. As research advances, further refinement of methylation panels and classification algorithms will enhance both detection sensitivity and accuracy of tissue of origin prediction, ultimately enabling earlier cancer diagnosis and improved patient outcomes.
Monitoring Treatment Response and Minimal Residual Disease (MRD) in Clinical Trials
This application note provides a detailed protocol for using whole-genome sequencing (WGS)-informed multiplex droplet digital PCR (ddPCR) to monitor treatment response and minimal residual disease (MRD) in B-cell lymphoma patients via circulating tumor DNA (ctDNA) analysis. The approach leverages patient-specific structural variants (SVs) and single nucleotide variants (SNVs) to achieve ultra-sensitive detection of residual disease, enabling early relapse identification and dynamic response assessment during clinical trials.
Key Clinical Validation Data
Table 1: Clinical Performance of WGS-Informed Multiplex ddPCR for MRD Detection
| Performance Metric | Result | Clinical Context |
|---|---|---|
| Detection Sensitivity | 0.0025% for SVs; 0.02% for SNVs/Indels | Limit of detection for patient-specific assays [41] |
| Baseline Detection Rate | 88% (7/8 patients) | ctDNA positivity in plasma at initial diagnosis [41] |
| Correlation with Imaging | 100% concordance at End-of-Treatment | ctDNA negativity correlated with PET-CT negative status post-primary treatment [41] |
| Early Relapse Detection | 25 weeks lead time | ctDNA detected in follow-up plasma 25 weeks prior to clinical manifestation of relapse [41] |
| Post-Cycle 1 Clearance | 50% (3/6 patients) | Clearance of ctDNA after a single cycle of primary treatment [41] |
The detection of Minimal Residual Disease (MRD) is a critical challenge in oncology clinical trials, as it allows for the precise assessment of treatment efficacy and the identification of patients at high risk of relapse. Circulating tumor DNA (ctDNA) has emerged as a powerful, non-invasive biomarker for MRD monitoring. This approach, often termed "liquid biopsy," enables real-time tracking of tumor dynamics through serial blood sampling.
Multiplex ddPCR represents a significant technological advancement for ctDNA analysis, offering absolute quantification of nucleic acids without the need for standard curves. By partitioning a sample into thousands of nanoliter-sized droplets, ddPCR allows for the detection of rare mutant alleles within a background of wild-type DNA with exceptional sensitivity and precision [42]. When informed by prior WGS of the tumor, multiplex ddPCR assays can be designed to track multiple patient-specific mutations simultaneously, thereby enhancing the robustness and sensitivity of MRD detection [41]. This protocol details the application of this integrated approach for monitoring patients in a clinical trial setting.
Experimental Protocol
Patient Enrollment and Sample Collection
- Patient Cohort: Patients with newly diagnosed B-cell lymphoma (e.g., Diffuse Large B-Cell Lymphoma, Follicular Lymphoma) are enrolled following informed consent [41].
- Baseline Sample Collection:
- Tumor Tissue: Collect a fresh-frozen tumor tissue biopsy.
- Matched Germline Control: Draw peripheral blood in EDTA tubes for extraction of germline genomic DNA.
- Longitudinal Liquid Biopsy Collection:
- Collect serial peripheral blood samples at critical clinical timepoints:
- At diagnosis
- After the first cycle of primary treatment
- At interim evaluation
- At final end-of-treatment evaluation
- During long-term follow-up (e.g., annually for 2 years)
- In case of suspected relapse: before treatment, at interim, and post-treatment [41]
- Use cell-free DNA BCT tubes (Streck) for blood collection. Process samples within 7 days by double centrifugation (1,600 ×g for 10 min, then 16,000 ×g for 10 min at 4°C) to isolate cell-free plasma. Aliquot and store plasma at -80°C [41].
- Collect serial peripheral blood samples at critical clinical timepoints:
Whole-Genome Sequencing and Bioinformatic Analysis
- DNA Extraction: Extract high-molecular-weight genomic DNA from paired tumor and normal specimens.
- Library Preparation and Sequencing: Prepare libraries (e.g., using TruSeq DNA PCR-Free kit, Illumina) and perform 30X whole-genome sequencing on both samples [41].
- Variant Calling:
- Identify patient-specific single nucleotide variants (SNVs) and indels. In validation studies, an average of 164 SNVs/indels per patient were identified, including mutations in key genes like
KMT2D,PIM1,SOCS1, andBCL2[41]. - Identify patient-specific structural variants (SVs), such as recurrent translocations including
t(14;18)(q32;q21)(IGH::BCL2) [41]. - Prioritize clonal, high-allele-frequency variants for assay design.
- Identify patient-specific single nucleotide variants (SNVs) and indels. In validation studies, an average of 164 SNVs/indels per patient were identified, including mutations in key genes like
Design and Validation of Patient-Specific Multiplex ddPCR Assays
- Assay Design: Design TaqMan probe-based ddPCR assays for the top 2-4 patient-specific SNVs/indels and 1-2 SVs.
- Multiplexing: Combine these assays into a single multiplex (m-ddPCR) reaction. Fluorophores (e.g., FAM, HEX/VIC) must be selected based on the detection channels of the dPCR platform [41] [43].
- Assay Validation:
- Test assays on diagnostic tumor DNA diluted into wild-type DNA.
- Establish analytical sensitivity and specificity. The referenced protocol achieved a detection sensitivity of 0.0025% for SV assays and 0.02% for SNVs/indel assays [41].
Cell-free DNA Extraction and Quantification
- Extraction: Extract cfDNA from 2-8 mL of plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen) on a vacuum manifold, eluting in 40 μL AVE buffer [41].
- Quality Control (QC): It is critical to assess cfDNA quality and quantity.
- Quantity: Use fluorometry or a ddPCR assay targeting a reference gene (e.g.,
STAT6) for absolute quantification [44]. - Fragment Size/Quality: Employ a multiplex ddPCR QC assay that amplifies targets of different lengths (e.g., 65 bp and 250 bp amplicons of the
EMC7gene) to assess fragmentation and rule out genomic DNA contamination [6]. - Inhibition Test: Include a spike-in control to detect PCR inhibitors [6].
- Quantity: Use fluorometry or a ddPCR assay targeting a reference gene (e.g.,
Multiplex ddPCR Setup and Data Analysis
- Reaction Setup: Prepare ddPCR reactions according to the manufacturer's instructions for your platform (e.g., Bio-Rad QX200, Qiagen QIAcuity, or Stilla naica).
- Partitioning and Amplification:
- Generate droplets or partitions.
- Perform PCR amplification with a thermocycling protocol optimized for the specific assays.
- Endpoint Reading and Analysis:
- Read partitions on an appropriate droplet reader (e.g., QX200 Droplet Reader) or imaging system (e.g., naica Prism3) [43].
- Set fluorescence amplitude thresholds to distinguish positive and negative droplets for each channel.
- Calculate ctDNA concentration: The concentration (copies/μL) is calculated by the instrument's software using Poisson statistics. Report as variant allele frequency (VAF) or mean tumor molecules per mL plasma [41] [15].
Diagram 1: Experimental workflow for WGS-informed multiplex ddPCR MRD monitoring.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for WGS-Informed Multiplex ddPCR
| Item | Function/Description | Example Product/Catalog Number |
|---|---|---|
| cfDNA Collection Tubes | Stabilizes cell-free DNA in blood samples for extended periods at room temperature. | Cell-Free DNA BCT Tubes (Streck) [41] |
| cfDNA Extraction Kit | Isulates and purifies cell-free DNA from plasma samples. | QIAamp Circulating Nucleic Acid Kit (Qiagen) [41] [15] |
| WGS Library Prep Kit | Prepares genomic DNA from tissue for Whole Genome Sequencing. | TruSeq DNA PCR-Free Library Prep Kit (Illumina) [41] |
| dPCR Supermix | Optimized reaction mix for digital PCR, including DNA polymerase, dNTPs, and buffers. | ddPCR Supermix for Probes (Bio-Rad) |
| Custom TaqMan Assays | Patient-specific primers and fluorescently-labeled probes for targeting SNVs/indels and SVs. | Custom TaqMan SNP Genotyping Assays (Thermo Fisher) [15] |
| Droplet Generation Oil | Immiscible oil for creating stable water-in-oil emulsion droplets during partition generation. | Droplet Generation Oil for Probes (Bio-Rad) |
| Reference Assay | Assay for a diploid gene to quantify total cfDNA concentration and assess sample quality. | EMC7 (65bp/250bp) or STAT6 assays [44] [6] |
Comparative Performance Data
Single-Target vs. Multitarget Detection Strategies
The choice between tracking a single mutation versus multiple mutations is a key consideration in MRD assay design. A comparative study in colorectal cancer (CRC) provides empirical data on the performance of both approaches.
Table 3: Single-Target (ST) ddPCR vs. Multitarget (MT) NGS in Postoperative CRC Monitoring
| Comparison Metric | Single-Target ddPCR | Multitarget NGS (16-plex) |
|---|---|---|
| Overall Concordance | 90% (Cohen's Kappa = 0.79) | 90% (Cohen's Kappa = 0.79) [15] |
| Preoperative Detection | 78% (72/92 samples) | 96% (88/92 samples) [15] |
| Postoperative Detection (Recurrence) | 50% (11/22 samples) | 45% (10/22 samples) [15] |
| Lead Time to Radiology | 4.0 months | 4.1 months [15] |
| Quantification Correlation | Pearson r = 0.985 | Pearson r = 0.985 [15] |
The data demonstrates that while MT approaches may offer higher sensitivity in a preoperative, high-disease-burden setting, both ST and MT strategies show remarkably similar performance in the postoperative MRD setting, where tumor burden is lowest. This suggests that a well-chosen single target can be as effective as a more complex MT approach for recurrence-risk stratification, potentially simplifying clinical trial assays [15].
Comparison of dPCR Platforms
Different dPCR technologies are available, and their performance can vary.
Table 4: Selected Commercial dPCR Platforms for ctDNA Analysis
| Platform (Brand) | Partitioning Technology | Key Features | Detection Channels |
|---|---|---|---|
| QX200/ddPCR (Bio-Rad) | Droplet (Oil-in-Water Emulsion) | Established workflow, high partition count | 2 (FAM, HEX/VIC) [45] |
| QIAcuity (Qiagen) | Solid-chip (Nanofluidic Microchambers) | Integrated partitioning, cycling, and imaging; no droplet transfer | Up to 5 channels [42] [45] |
| naica (Stilla) | Crystal Digital PCR (2D Droplet Array) | 3-color multiplexing, high-resolution imaging | 3 to 6 channels [43] |
| QuantStudio Absolute Q (Thermo Fisher) | Solid-chip (Microchambers) | Fully integrated digital PCR system; walkaway automation | 4 analysis channels [42] |
A study comparing a droplet-based system (Bio-Rad ddPCR) with a solid-state system (Qiagen QIAcuity) for analyzing lung and colorectal cancer liquid biopsy samples found a moderate agreement between the platforms. However, the solid dPCR system demonstrated a higher sensitivity in detecting mutated cases, particularly for EGFR mutations in NSCLC (100% vs. 58.8% detection compared to tissue) [45]. This highlights the importance of platform selection and validation.
Troubleshooting and Technical Notes
- Low ctDNA Yield: Ensure rapid processing of blood samples and use validated cfDNA collection tubes. Increase plasma input volume for extraction if possible.
- Poor Droplet Resolution or Partition Quality: Verify oil and sample storage conditions. Ensure proper droplet generation technique and equipment maintenance.
- Inconsistent Background Signal: Titrate probe concentrations and optimize annealing temperature during assay validation. Include multiple negative controls.
- False Positives/Negatives: Apply stringent, pre-defined thresholds for calling a sample positive. For MT-NGS approaches, a common rule is to require ≥2 variant calls above background [15]. Always confirm results with repeated testing if sample volume allows.
- Assay Dropout: Tracking multiple, clonal variants in a multiplex format mitigates the risk of missing MRD due to clonal evolution or assay failure [41].
Longitudinal Disease Monitoring and Tracking Tumor Evolution
Circulating tumor DNA (ctDNA) analysis has emerged as a transformative tool in oncology, enabling non-invasive, real-time monitoring of tumor dynamics and evolutionary trajectories. The analysis of ctDNA, a fraction of cell-free DNA (cfDNA) shed into the bloodstream by apoptotic and necrotic tumor cells, provides a critical window into tumor heterogeneity and adaptive changes under therapeutic pressure [46]. For researchers and drug development professionals, leveraging ctDNA through sensitive detection platforms like multiplex droplet digital PCR (ddPCR) offers unprecedented opportunities to decipher cancer evolution, assess treatment efficacy, and identify emergent resistance mechanisms.
This application note details the integration of multiplex ddPCR assays within a longitudinal disease monitoring framework, providing validated protocols and analytical frameworks to track tumor evolution across the cancer disease course. The content is structured to equip researchers with practical methodologies aligned with consortium-established validation standards [47] [48], enabling robust implementation in both basic research and translational drug development settings.
The Role of ctDNA in Tracking Tumor Evolution
Tumor evolution is characterized by the dynamic selection of cellular subpopulations with distinct genomic alterations, leading to intratumour heterogeneity [49]. Longitudinal ctDNA analysis captures this spatial and temporal heterogeneity non-invasively by profiling DNA fragments derived from all metastatic sites, overcoming the limitations of single-site tissue biopsies [49] [46]. The short half-life of ctDNA (approximately 2 hours) enables near real-time assessment of tumor burden and genomic changes, making it an ideal biomarker for monitoring disease progression and treatment response [6] [46].
In advanced cancers, ctDNA levels generally correlate with tumor burden, and changes in these levels can predict radiographic response to therapy earlier than conventional imaging [27]. Furthermore, the evolution of resistance mutations can be detected in plasma weeks before clinical evidence of disease progression, creating a critical window for therapeutic intervention [27] [46]. For drug development professionals, this capability enables more efficient assessment of treatment efficacy and resistance mechanisms during clinical trials.
Multiplex ddPCR for ctDNA Analysis: Advantages and Technical Considerations
Droplet digital PCR represents a highly sensitive, absolute quantification method for detecting rare mutant alleles in a background of wild-type DNA. Multiplex ddPCR further enhances this capability by simultaneously tracking multiple tumor-specific alterations from limited patient samples, making it particularly suited for longitudinal monitoring studies.
- High Sensitivity and Specificity: ddPCR detects somatic alterations at very low variant allele frequencies (VAFs), with studies reporting reliable detection down to 0.01% VAF [23]. This sensitivity is crucial for detecting minimal residual disease (MRD) and early relapse in patients with solid tumors.
- Cost-Effectiveness: Compared to next-generation sequencing (NGS), ddPCR offers significantly lower operational costs, with studies indicating a 5–8.5-fold reduction in expenses [23]. This makes it economically viable for high-frequency monitoring throughout a patient's treatment journey.
- Quantitative Precision: ddPCR provides absolute quantification of mutant DNA molecules without relying on calibration curves, enabling precise measurement of ctDNA concentration changes over time—a critical requirement for assessing tumor dynamics [23] [6].
Table 1: Comparison of ctDNA Detection Platforms
| Feature | Multiplex ddPCR | Targeted NGS Panels |
|---|---|---|
| Sensitivity | High (VAF ~0.01%) [23] | Moderate (VAF ~0.1%+) [23] |
| Multiplexing Capability | Moderate (Typically 2-6 targets) | High (Hundreds of genes) |
| Cost per Sample | Low [23] | High |
| Turnaround Time | Fast (Hours) | Slow (Days to Weeks) |
| Quantitative Nature | Absolute quantification | Relative quantification |
| Ideal Application | Tracking known mutations over time | Discovering novel alterations |
Experimental Protocols for Longitudinal ctDNA Monitoring
Pre-Analytical Phase: Blood Collection and Plasma Processing
Critical Step: Standardized blood collection and processing is essential to prevent genomic DNA contamination from white blood cell lysis, which can dramatically dilute the ctDNA fraction [48] [46].
- Blood Collection: Draw patient blood into Streck Cell-Free DNA BCT tubes or similar specialized collection tubes containing additives that stabilize blood cells. These tubes prevent cell lysis during transport and storage [23] [46].
- Plasma Separation: Process blood samples within 4-6 hours of collection. Centrifuge tubes at 2,000 × g for 10 minutes at room temperature to separate plasma from cellular components.
- Secondary Centrifugation: Transfer the supernatant (plasma) to a fresh tube and perform a second centrifugation at 10,000 × g for 10 minutes to remove any remaining cellular debris [6].
- Storage: Aliquot the clarified plasma and store at -80°C until cfDNA extraction. Avoid repeated freeze-thaw cycles.
Analytical Phase: ctDNA Extraction and Multiplex ddPCR Assay
Objective: Isolate high-quality cfDNA and detect tumor-specific mutations with high sensitivity and specificity.
- cfDNA Extraction: Extract cfDNA from 2-4 mL of plasma using commercially available kits (e.g., QIAsymphony DSP Circulating DNA Kit). Elute DNA in a small volume (e.g., 60 µL) of elution buffer to maximize concentration [6].
- Assay Design (Tumor-Informed):
- Perform initial sequencing of the patient's tumor tissue (e.g., using a targeted NGS panel like the Ion AmpliSeq Cancer Hotspot Panel v2) to identify 1-2 somatic mutations with the highest variant allele frequencies [23].
- Design custom ddPCR assays (FAM/HEX probes) for these patient-specific mutations.
- Multiplex ddPCR Workflow:
- Concentrate the extracted cfDNA using centrifugal filters (e.g., Amicon Ultra-0.5) if necessary.
- Prepare the ddPCR reaction mix containing the DNA template, multiplexed mutation-specific probes, and ddPCR Supermix.
- Generate droplets using an Automated Droplet Generator or QX200 Droplet Generator. Typically, 20,000 droplets per sample are generated.
- Perform endpoint PCR amplification with the following cycling conditions: 95°C for 10 minutes (enzyme activation), followed by 40 cycles of 94°C for 30 seconds (denaturation) and 55-60°C for 1 minute (annealing/extension), and a final 98°C step for 10 minutes (enzyme deactivation). A ramp rate of 2°C/second is recommended.
- Read the plate on a droplet reader (e.g., QX200 Droplet Reader) which counts the number of positive (mutant) and negative (wild-type) droplets in each channel.
- Data Analysis:
- Use the manufacturer's software (e.g., QuantaSoft) to calculate the concentration of mutant DNA (copies/µL) and variant allele frequency (VAF) using Poisson statistics.
- The threshold for ctDNA positivity should be established using negative controls (plasma from healthy donors) and may be defined by a minimum number of mutant droplets (e.g., ≥ 3) above the background noise [23] [6].
Post-Analytical Phase: Data Interpretation and Longitudinal Tracking
Objective: Translate ddPCR results into meaningful biological insights about tumor evolution.
- Quantitative Trend Analysis: Plot the concentration of mutant DNA (copies/mL of plasma) or VAF against the timeline of sample collection and therapeutic interventions.
- Correlation with Clinical Parameters: Integrate ctDNA data with radiographic imaging (CT, MRI), serum tumor markers (e.g., CEA), and clinical assessment of symptoms.
- Defining Molecular Response: A significant decrease (e.g., >50% or >90%) in ctDNA levels after treatment initiation may be classified as a molecular response, often preceding radiographic changes [27] [46].
- Identifying Resistance: The emergence of new mutant populations (detectable by designing probes for known resistance mutations, e.g., EGFR T790M) in subsequent plasma samples indicates clonal evolution and potential therapy resistance [27].
Visualizing Workflows and Tumor Evolution
The following diagram illustrates the complete workflow for longitudinal monitoring of tumor evolution using multiplex ddPCR.
The following diagram conceptualizes the principles of tumor evolution and how it is reflected in ctDNA dynamics under therapeutic pressure.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for Multiplex ddPCR ctDNA Analysis
| Item | Function | Example Products/Assays |
|---|---|---|
| Cell-Free DNA Collection Tubes | Stabilizes nucleated blood cells for up to 14 days, preventing gDNA contamination and preserving ctDNA profile. | Streck Cell-Free DNA BCT tubes, PAXgene Blood ccfDNA Tubes [23] [46] |
| cfDNA Extraction Kits | Isolate short-fragment cfDNA from plasma with high efficiency and reproducibility. | QIAsymphony DSP Circulating DNA Kit (Qiagen) [6] |
| Droplet Digital PCR System | Platform for partitioning samples into nanoliter droplets, PCR amplification, and absolute quantification of target molecules. | Bio-Rad QX200 system [23] [6] |
| Custom ddPCR Assays | Fluorescence-labeled probes (FAM/HEX) designed to specifically detect patient- or tumor-specific point mutations. | Bio-Rad ddPCR Mutation Assays, Custom TaqMan Assays [23] |
| NGS Hotspot Panel | For initial tumor tissue genotyping to identify clonal mutations for patient-specific ddPCR assay design. | Ion AmpliSeq Cancer Hotspot Panel v2 (Thermo Fisher) [23] |
| Bisulfite Conversion Kit | (For methylation assays) Chemically converts unmethylated cytosines to uracils, allowing detection of methylation status. | EZ DNA Methylation-Lightning Kit (Zymo Research) [6] |
Longitudinal monitoring of ctDNA using multiplex ddPCR provides a powerful, sensitive, and cost-effective strategy for tracking tumor evolution in real time. The protocols and frameworks outlined in this application note, grounded in consortium best practices and recent clinical research, provide a roadmap for researchers and drug development scientists to implement this technology effectively. By capturing the dynamic genomic landscape of tumors, this approach enables deeper insights into cancer biology, therapy response, and resistance mechanisms, ultimately accelerating the development of more effective, personalized cancer treatments. Future directions will involve standardizing these protocols across laboratories and integrating ctDNA data with other multi-omic platforms to build a more comprehensive understanding of cancer evolution.
Optimizing Performance and Overcoming Technical Challenges in Multiplex ddPCR
The analysis of circulating tumor DNA (ctDNA) via multiplex droplet digital PCR (ddPCR) represents a transformative approach in precision oncology, enabling non-invasive tumor genotyping, therapy monitoring, and minimal residual disease (MRD) detection. A paramount challenge in this field, especially when targeting low-frequency variants in early-stage cancer, is the accurate distinction of true positive signals from false positives. False positives can arise from various sources, including PCR errors, sample cross-contamination, and inadequate bioinformatic filtering, potentially leading to incorrect clinical interpretations. This Application Note details proven, practical strategies for robust multiplex ddPCR assay design and analytical threshold setting to enhance the reliability of ctDNA detection for research and drug development applications.
Technical Strategies for False Positive Minimization
Harnessing Unique Molecular Identifiers (UMIs) and Duplex Sequencing
The implementation of Unique Molecular Identifiers (UMIs) is a critical first step in mitigating false positives originating from amplification artifacts. UMIs are short random nucleotide sequences ligated to individual DNA molecules before PCR amplification. This allows bioinformatic tracing of each sequenced read back to its original template, enabling the subtraction of errors introduced in later PCR cycles [50].
A more advanced application of this principle is duplex sequencing, which uses both strands of a DNA molecule for error correction. In this method, mutations are only considered valid if they are detected in both complementary strands originating from the same original molecule. This approach dramatically reduces false positives caused by polymerase errors or DNA damage, as these events are unlikely to occur at the same genomic position on both strands. Optimized library preparation kits are available that support this workflow, significantly improving signal-to-noise ratios in ctDNA analysis [51].
Establishing a Dynamic Limit of Detection (LoD)
A fixed analytical threshold may be insufficient for ctDNA analysis due to varying tumor DNA fractions. Implementing a dynamic Limit of Detection (LoD) calibrated to sequencing depth and input DNA quality offers a more reliable strategy. The required depth of coverage (DoC) increases exponentially for detecting lower variant allele frequencies (VAFs); achieving a 99% probability of detecting a variant at 0.1% VAF requires approximately 10,000x coverage [50].
Table 1: Coverage Requirements for Variant Detection at 99% Probability
| Variant Allele Frequency (VAF) | Required Depth of Coverage |
|---|---|
| 1.0% | 1,000x |
| 0.5% | 2,000x |
| 0.1% | ~10,000x |
This dynamic approach ensures that the LoD is continually adjusted based on sample-specific parameters, thereby enhancing result reliability and confidence in clinical interpretation [50].
Implementing Bioinformatics "Allowed" and "Blocked" Lists
Strategic bioinformatics pipelines can further enhance accuracy. Utilizing "allowed" and "blocked" lists for variant calling helps filter out commonly occurring technical artifacts or polymorphisms. An "allowed" list comprises known, validated mutations of clinical interest, while a "blocked" list includes recurrent sequencing errors or benign polymorphisms. This pre-filtering step minimizes the reporting of false positive variants without compromising the detection of true tumor-derived mutations [50].
Optimizing Multiplex ddPCR Assay Design
In the context of ddPCR, careful multiplex assay optimization is crucial. This involves:
- Probe and Primer Optimization: Validating individual primer and probe sets in singleplex reactions before combining them into multiplex assays. This includes finding optimal concentrations, annealing temperatures, and checking for primer-dimer formations [52].
- Fluorophore Selection and Cross-Talk Correction: Using fluorophores with distinct emission spectra and applying custom cross-talk matrices in the analysis software to correct for signal bleed-through between channels. Advanced dPCR systems with multiple detection channels facilitate high-order multiplexing [52].
- Threshold Setting with Wild-Type Controls: Using wild-type-only (WT-only) control samples to establish a baseline fluorescence amplitude and determine the threshold for positive droplet calling. This helps estimate the false-positive rate for each assay, allowing for the setting of a minimum number of mutant-positive droplets required to call a sample positive [24].
Experimental Protocols
Protocol: UMI-Based Library Preparation for ctDNA
This protocol is adapted for ctDNA analysis from plasma-derived cell-free DNA (cfDNA) [50] [51].
- cfDNA Extraction: Extract cfDNA from 2-10 mL of patient plasma using a specialized circulating nucleic acid kit. Elute in a suitable buffer and quantify using a fluorometer.
- End-Repair and A-Tailing: Perform enzymatic end-repair and dA-tailing of the fragmented cfDNA to create blunt-ended, 5'-phosphorylated DNA fragments compatible with adapter ligation.
- UMI-Adapter Ligation: Ligate double-stranded adapters containing unique molecular identifiers (UMIs) to the prepared DNA fragments. Use a high-conversion efficiency ligation enzyme mix to maximize the yield of usable fragments.
- Library Amplification: Amplify the ligated library using a high-fidelity DNA polymerase for a limited number of PCR cycles (e.g., 10-14 cycles) to minimize amplification errors.
- Target Enrichment (for NGS): For NGS workflows, hybridize the library to a target-specific panel (e.g., a cancer gene panel) to enrich for genomic regions of interest. For ddPCR, this step is not required.
- Purification and Quantification: Purify the final library and quantify it before sequencing or ddPCR analysis.
Protocol: Setting Analytical Thresholds with Wild-Type Controls
This procedure outlines how to empirically determine false-positive rates and set robust thresholds for ddPCR [24].
- Assay Design: Design and validate mutation-specific ddPCR assays (e.g., for TP53 or PIK3CA mutations).
- Wild-Type Control Preparation: Isolate plasma DNA from at least five healthy donor samples using the same cfDNA extraction kit and protocol used for patient samples.
- ddPCR Run: Run the wild-type control samples alongside no-template controls (NTCs) using the same mutant-specific ddPCR assay conditions intended for patient samples.
- False-Positive Rate Calculation: Analyze the wild-type control wells. Count the total number of mutant-positive droplets detected across all wild-type replicates. The false-positive rate is calculated based on this count relative to the total number of droplets analyzed.
- Threshold Definition: Establish a minimum threshold for mutant copies (or positive droplets) required to call a patient sample positive. This threshold should be set significantly above the background level observed in wild-type controls (e.g., >3 standard deviations above the mean of the wild-type signals). For example, if a series of wild-type controls yields 0 or 1 false-positive mutant droplet, the threshold for a true positive can be set to >1 or >2 mutant-positive droplets [24].
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Robust Multiplex ddPCR
| Reagent / Solution | Function | Key Considerations |
|---|---|---|
| High-Fidelity DNA Polymerase | Amplifies target DNA sequences with minimal errors during PCR. | Reduces introduction of polymerase-based mutations that can be misinterpreted as low-frequency variants [51]. |
| UMI-Adapters | Uniquely tags original DNA molecules before amplification. | Enables bioinformatic correction of amplification errors and deduplication for accurate quantification of original templates [50]. |
| Multiplex ddPCR Probe Master Mix | Provides optimized buffer and nucleotides for probe-based multiplex ddPCR. | Ensures efficient amplification and clear fluorescence signal separation in multi-target reactions [52]. |
| Target-Specific Fluorescent Probes | Detects and differentiates multiple genetic targets in a single reaction. | Probes for different targets must be conjugated to fluorophores with non-overlapping emission spectra [52]. |
| Wild-Type Control DNA | Serves as a negative control for estimating assay-specific false-positive rates. | Essential for establishing a baseline signal and defining the analytical threshold for variant calling [24]. |
Workflow and Pathway Diagrams
The following diagram illustrates the core experimental workflow for a robust multiplex ddPCR assay, integrating key strategies for minimizing false positives.
This workflow highlights the two critical phases where false positives are most effectively controlled: during wet-lab preparation (UMI ligation) and dry-lab analysis (bioinformatic filtering).
The analysis of circulating tumor DNA (ctDNA) via liquid biopsy represents a transformative approach in oncology, enabling non-invasive tumor genotyping, monitoring of treatment response, and detection of minimal residual disease [9]. Within this field, droplet digital PCR (ddPCR) has emerged as a premier technology for the quantification of low-abundance mutations due to its exceptional sensitivity and absolute quantification capabilities without the need for standard curves [16] [53]. However, the detection of ctDNA is analytically challenging as it often constitutes less than 0.1% of the total cell-free DNA in early-stage cancers, necessitating exquisitely optimized assays [9] [53]. This application note provides a detailed framework for optimizing two critical parameters in multiplex ddPCR assay development: primer/probe concentrations and thermal cycling conditions, specifically within the context of ctDNA analysis for cancer research and drug development.
Primer and Probe Concentration Optimization
Optimal primer and probe concentrations are fundamental to achieving high signal-to-noise ratios, specifically minimizing false-positive signals while retaining robust detection of true mutant alleles. The following table summarizes recommended concentration ranges and optimization strategies based on recent ctDNA studies.
Table 1: Optimization of Primer and Probe Concentrations for ddPCR Assays
| Component | Concentration Range | Optimization Strategy | Impact on Assay Performance |
|---|---|---|---|
| Primers | 0.05 - 1.0 µM [54] [55] | • Start at 0.5 µM and titrate in 0.1 µM increments.• Ensure primer pairs have Tm within 5°C of each other [55]. | Higher concentrations may increase spurious amplification; lower concentrations reduce sensitivity [54]. |
| Hydrolysis Probes | 50 - 500 nM [16] | • Titrate alongside primers.• Use LNA or mediator probes to enhance specificity for point mutations [16] [56]. | Reduces background fluorescence and improves cluster separation in 2D amplitude plots [16]. |
| Multiplex Probes | 100 - 300 nM [56] | • Standardize concentrations across all assays in the panel.• For mediator probe PCR, use 240 nM universal reporter [56]. | Ensures balanced fluorescence amplitudes for different targets, enabling accurate multiplexing [56]. |
Key Experimental Protocol: Titration of Primer and Probe Concentrations
- Preparation: Prepare a master mix containing 11 µL of 2x ddPCR Supermix for Probes (no dUTP), 1 µL of template DNA (e.g., a synthetic DNA fragment or gBlock containing the target mutation at ~100 copies), and nuclease-free water [16].
- Reaction Setup: Aliquot the master mix into separate tubes. Prepare a series of reactions varying the forward and reverse primer concentrations (e.g., 0.1, 0.25, 0.5, and 0.75 µM) while maintaining a constant probe concentration (e.g., 250 nM). In a parallel series, vary the probe concentration (e.g., 100, 200, 300, 400 nM) with constant primer concentrations [16] [24].
- Droplet Generation and PCR: Generate droplets using the QX200 AutoDG system. Perform PCR amplification with a standardized cycling protocol: 95°C for 10 min (1 cycle); 94°C for 30 s and a universal 55-60°C for 60 s (45-55 cycles); and a final hold at 12°C [16] [24].
- Analysis: Read the plate on a QX200 droplet reader. Analyze results using the manufacturer's software (e.g., QuantaSoft). The optimal concentration is identified by the combination that yields the highest number of positive droplets for the target mutation, the clearest separation between positive and negative droplet clusters, and the lowest false-positive rate in no-template and wild-type-only controls [24].
Thermal Cycling Parameter Optimization
Precise thermal cycling is critical for efficient amplification, especially in multiplex assays where several primer/probe sets must function simultaneously under identical conditions. The following table outlines key parameters and their optimization.
Table 2: Optimization of Thermal Cycling Parameters for ddPCR
| Parameter | Recommended Conditions | Optimization Strategy | Impact on Assay Performance |
|---|---|---|---|
| Initial Denaturation | 94–98°C for 1–3 min [57] | • Use higher temperatures (98°C) for GC-rich templates [57].• This step also activates hot-start DNA polymerases. | Ensures complete separation of DNA strands; higher temperatures can help inactivate nucleases [57]. |
| Denaturation | 94–98°C for 15–30 s [57] [54] | • Standardize at 94°C for 30 s in most cases. | Inadequate denaturation leads to a drastic reduction in PCR yield [57]. |
| Annealing Temperature | 50–65°C for 15–60 s [57] [54] | • Start 3–5°C below the lowest primer Tm [57].• Use a thermal gradient (e.g., 50–65°C) to determine the temperature that gives the highest fluorescence amplitude and best cluster separation [24]. | Too low: non-specific binding and false positives. Too high: reduced yield and sensitivity [57]. A universal temperature of 54–60°C can be achieved with isostabilizing buffers [57] [56]. |
| Extension | 68–72°C for 15–60 s/kb [57] [54] | • Typically combined with annealing in a two-step protocol if the annealing temperature is within 3°C of extension [57]. | Essential for polymerase activity; insufficient time leads to incomplete amplicons [57]. |
| Cycle Number | 40–55 cycles [16] [24] | • 40 cycles is standard; increase to 45–55 for very low-abundance targets (<0.1% AF) [16]. | Higher cycles enhance sensitivity for low-concentration targets but may increase background [16]. |
Key Experimental Protocol: Annealing Temperature Gradient
- Reaction Setup: Prepare a master mix containing all reaction components—supermix, optimized primer/probe concentrations, and a template DNA known to be positive for the target mutation(s) at a low allele frequency (e.g., 1%) [24].
- Thermal Cycling: Program the thermal cycler with a gradient across the block for the annealing step. A recommended range is 50°C to 65°C, covering the calculated Tm of the primers [57] [55].
- Analysis: After droplet reading, assess the results from each temperature. The optimal annealing temperature is identified by the clearest discrimination between positive and negative droplet populations, the highest number of mutant-positive droplets, and the absence of rain (droplets with intermediate fluorescence) [24]. For multiplex assays, the chosen temperature must provide optimal performance for all assays simultaneously [16] [56].
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful development of a multiplex ddPCR assay requires a suite of specialized reagents and controls. The following table details key solutions for ctDNA research.
Table 3: Essential Reagents and Materials for ctDNA ddPCR Assay Development
| Reagent/Material | Function and Importance | Example Products & Notes |
|---|---|---|
| ddPCR Supermix | Provides the core components for PCR, including a DNA polymerase with high fidelity and stability. | ddPCR Supermix for Probes (no dUTP) from Bio-Rad [16]. The "no dUTP" formulation is crucial to prevent carryover contamination. |
| Synthetic DNA Controls | Serve as well-defined positive controls for assay validation and determining limits of detection (LOD). | gBlock Gene Fragments (IDT) [16] [56]. These are sequence-verified double-stranded DNA fragments. |
| Reference Genomic DNA | Serves as a wild-type control to establish baseline and false-positive rates. | Human genomic DNA from commercial vendors (e.g., Promega, Roche) [16] [56]. |
| Cell-Free DNA Extraction Kits | Isolate cfDNA from plasma with high efficiency and reproducibility, critical for quantitative accuracy. | QIAamp Circulating Nucleic Acid Kit (Qiagen) [16] [24], or Maxwell RSC ccfDNA Plasma Kit (Promega) [16]. |
| Locked Nucleic Acid (LNA) Probes | Enhance hybridization specificity and allelic discrimination, especially for single-base mutations. | PrimeTime LNA probes (IDT) [16]. Incorporation of LNA bases increases the Tm and improves mismatch discrimination [16]. |
| Mediator Probes / Universal Reporters | Enable optimization-free multiplexing by decoupling target detection from signal generation. | A system using label-free mediator probes and standardized fluorogenic universal reporters [56]. Allows use of identical conditions for different targets. |
Workflow and Pathway Diagrams
The following diagram illustrates the complete workflow for developing and optimizing a multiplex ddPCR assay for ctDNA analysis, from initial design to data analysis.
Figure 1: Sequential workflow for ctDNA ddPCR assay development.
The optimization of primer/probe concentrations and thermal cycling parameters forms a critical signaling pathway within the ddPCR system that directly determines the reliability of ctDNA detection, as visualized below.
Figure 2: Logical relationship between optimization parameters and assay performance.
The rigorous optimization of primer/probe concentrations and thermal cycling parameters is a prerequisite for generating robust, reliable, and clinically actionable data from multiplex ddPCR assays in ctDNA research. By following the detailed protocols and guidelines outlined in this document, researchers can develop highly sensitive and specific assays capable of detecting and quantifying rare mutant alleles in a high background of wild-type DNA. This level of performance is essential for advancing applications in early cancer detection, monitoring minimal residual disease, and guiding targeted therapy in oncology drug development.
The analysis of circulating tumor DNA (ctDNA) using multiplex droplet digital PCR (ddPCR) offers unprecedented potential for non-invasive cancer monitoring, treatment selection, and response assessment. However, the analytical success of these sophisticated applications is critically dependent on the management of pre-analytical variables that begin the moment blood is drawn from a patient. ctDNA presents unique challenges as a biomarker—it exists as short DNA fragments (typically 160-180 base pairs) in low abundance amidst a background of wild-type cell-free DNA (cfDNA) derived from normal cells, with ctDNA often constituting only 0.1% to 1% of total cfDNA in early-stage cancers [58]. This inherent biological scarcity means that improper pre-analytical handling can easily compromise sample integrity, leading to false-negative results or inaccurate variant allele fraction (VAF) quantification [59] [60]. Effective management of blood collection tubes, centrifugation parameters, and storage conditions therefore forms the foundational framework upon which reliable ctDNA analysis is built, particularly for the exacting requirements of multiplex ddPCR applications.
Blood Collection Tube Selection and Properties
The choice of blood collection tube represents the first critical decision point in the ctDNA workflow, as different additives and preservatives significantly impact sample stability and downstream analytical performance. Conventional EDTA tubes are widely available but require rapid sample processing (typically within 1-2 hours) to prevent white blood cell lysis and the consequent release of genomic DNA, which dilutes the already scarce ctDNA fraction [59] [60]. This limitation has driven the adoption of specialized blood collection tubes containing novel preservatives that stabilize blood samples for extended periods, facilitating transportation from clinical settings to testing laboratories.
Table 1: Blood Collection Tube Types for ctDNA Analysis
| Tube Type (Top Color) | Additives | Mechanism of Action | Stability/Processing Requirements | Primary Applications in ctDNA Research |
|---|---|---|---|---|
| K₂EDTA (Lavender/Pink) | Potassium EDTA | Chelates calcium ions to prevent clotting | Process within 1-2 days; significant WBC lysis after 1 day [59] [60] | Hematology, buffy coat preparation [61] |
| Cell-Free DNA BCT (Streck) | Proprietary preservative | Stabilizes nucleated blood cells, prevents lysis | Room temperature storage for up to 7-14 days [59] [16] | Preferred for ctDNA; enables sample transport [59] [16] |
| Citrate (Light Blue) | Sodium citrate (3.2%) | Weak calcium chelator | Requires timely processing | Coagulation studies [61] [62] |
| Heparin (Green) | Sodium/Lithium heparin | Inhibits thrombin formation | Not recommended for PCR; inhibits enzymatic reactions [63] | Chemistry panels, stat tests [61] |
| Serum Separator (Gold/Red) | Clot activator, gel separator | Promotes clotting, separates serum | 15-30 min clotting time; risk of WBC genomic DNA release [61] | Chemistry, serology; not ideal for ctDNA [61] |
For ctDNA analysis, plasma is strongly recommended over serum because serum preparation involves clotting, which can release genomic DNA from white blood cells and potentially dilute the ctDNA signal [58]. The implementation of preservative tubes such as Streck cell-free DNA BCT has demonstrated significant utility in ctDNA workflows, effectively maintaining sample integrity during transport and temporary storage by preventing white blood cell lysis and preserving the native fragmentomic profile of ctDNA [59] [16].
Centrifugation Protocols for Plasma Preparation
Centrifugation protocols for plasma preparation must achieve two competing objectives: complete removal of cellular components to prevent contamination from cellular genomic DNA, while simultaneously maximizing the recovery of the scarce ctDNA fraction. A standardized two-step centrifugation approach has emerged as the consensus method across multiple studies, with specific parameters optimized for ctDNA analysis [59] [58].
Table 2: Centrifugation Parameters for Plasma Preparation
| Centrifugation Step | Speed | Time | Temperature | Purpose | Key Considerations |
|---|---|---|---|---|---|
| Initial Spin | 1,600 × g | 10 minutes | 4°C [59] [58] | Separation of plasma from blood cells | Brake should be applied; generates platelet-poor plasma |
| Second Spin | 4,100 × g [59] OR 16,000 × g [58] | 10-15 minutes | 4°C [59] [58] | Removal of residual cells and debris | No significant difference in ccfDNA yield or ctDNA VAF between speeds [59] |
Recent rigorous investigations have demonstrated that a second centrifugation speed of 4,100 × g shows no significant difference in cfDNA yield or ctDNA variant allele fraction compared to higher-speed centrifugation at 16,000 × g [59]. This finding has important practical implications for clinical implementation, as the lower-speed protocol can be performed using standard clinical centrifuges rather than requiring specialized high-speed equipment that processes limited sample volumes. The addition of a third centrifugation step at similar parameters to the second spin has similarly shown no demonstrable benefit in reducing white blood cell contamination or improving ctDNA recovery, supporting the efficiency of the two-step protocol [59].
Detailed Experimental Protocol: Plasma Processing
Materials Required:
- Blood collected in Streck cell-free DNA BCT or K₂EDTA tubes
- Refrigerated centrifuge capable of 4,100 × g
- Sterile pipettes and aerosol-resistant tips
- Polypropylene transfer tubes
- Personal protective equipment (gloves, lab coat, safety goggles)
Procedure:
- Sample Acceptance: Upon receipt, record collection time and visually inspect tubes for cracks or clot formation.
- First Centrifugation: Transfer blood collection tubes to a refrigerated centrifuge (4°C). Balance tubes precisely. Centrifuge at 1,600 × g for 10 minutes with brake applied.
- Plasma Transfer: Using sterile pipettes, carefully transfer the upper plasma layer to clean polypropylene tubes, avoiding the buffy coat layer at all costs. Leave approximately 0.5 cm of plasma above the buffy coat to prevent cellular disturbance.
- Second Centrifugation: Centrifuge the transferred plasma at 4,100 × g for 15 minutes at 4°C.
- Final Plasma Collection: Transfer the supernatant to fresh tubes, again carefully avoiding the pellet that may form at the bottom of the tube.
- Aliquoting: Aliquot processed plasma into low DNA-binding tubes to avoid repeated freeze-thaw cycles in downstream applications.
Quality Control Notes:
- Process EDTA tubes within 2 hours of blood draw when using conventional collection methods.
- Streck BCT tubes can be processed within 7 days when stored at room temperature.
- Document any hemolysis observed, as this may affect downstream analysis.
Sample Storage and Stability
The stability of cfDNA samples throughout storage is a multi-factorial consideration encompassing temperature, duration, and sample state (whole blood vs. plasma vs. extracted DNA). When plasma is processed according to the optimized centrifugation protocols above, cfDNA remains stable at -80°C for extended periods, making this the recommended storage condition for plasma samples destined for ctDNA analysis [58] [60]. Comparative studies have investigated the impact of fresh versus frozen plasma on cfDNA yield with varying results—some quantification methods show higher yield from fresh plasma, while droplet digital PCR (ddPCR) has demonstrated higher yield from frozen plasma, though critically, no significant differences were observed in ctDNA variant allele fraction between fresh and frozen plasma [59].
For whole blood samples, the choice of collection tube dictates storage stability. Blood collected in conventional EDTA tubes demonstrates significant white blood cell lysis after approximately 1 day of storage, resulting in contaminating genomic DNA that dilutes the ctDNA fraction [59]. In contrast, preservative tubes such as Streck cell-free DNA BCT maintain sample integrity for up to 7-14 days at room temperature, facilitating sample transportation from collection sites to processing facilities [59] [16]. Extracted cfDNA samples should be stored in low DNA-binding tubes at -80°C, with aliquoting to minimize freeze-thaw cycles that can progressively fragment DNA and potentially impact fragmentation-based analytical methods [58].
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of ctDNA analysis requires specific specialized reagents and materials designed to maintain sample integrity and support downstream applications.
Table 3: Essential Research Reagent Solutions for ctDNA Analysis
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Cell-Free DNA BCT (Streck) | Stabilizes nucleated blood cells | Enables room temperature transport for up to 7-14 days; critical for multi-center trials [59] [16] |
| ccfDNA Plasma Extraction Kits | Isolate cfDNA from plasma | Use kits specifically designed for cfDNA; Promega Maxwell RSC, Qiagen QIAamp Circulating Nucleic Acid Kit [16] |
| DNA LoBind Tubes | Store cfDNA samples | Minimize DNA adhesion to tube walls [16] |
| gBlock Synthetic DNA | Spike-in control | XenT gBlock (IDT) monitors extraction efficiency; add prior to extraction [16] |
| Droplet Digital PCR Reagents | Absolute quantification of ctDNA | Bio-Rad ddPCR Supermix; requires optimization of primer/probe concentrations [16] |
| Bisulfite Conversion Kits | DNA methylation analysis | Required for methylation-based ddPCR assays; Zymo Research EZ DNA Methylation kit [7] |
Quality Assurance and Control Methods
Robust quality assurance practices are essential throughout the pre-analytical phase to ensure the reliability of downstream ctDNA analysis. The implementation of external synthetic DNA controls, such as XenT gBlocks spiked into plasma samples prior to nucleic acid extraction, provides a mechanism to quantify and monitor cfDNA extraction efficiency, enabling more accurate extrapolation of mutation levels in original patient samples [16]. For ddPCR applications, the RPP30 gene serves as an effective reference locus for quantifying total cfDNA content, as it is highly conserved, unique in the genome, and rarely impacted by copy number changes [16].
The quantification method selected for cfDNA analysis can yield substantially different results, highlighting the importance of methodological consistency. Fluorometric methods (e.g., Qubit) measure total DNA concentration but cannot distinguish ctDNA from wild-type cfDNA, while PCR-based approaches (e.g., ddPCR) provide absolute quantification of specific targets but require prior knowledge of mutations of interest [59]. The correlation between different quantification methods has shown variability across studies, reinforcing the need for consistent methodology within a study or clinical trial [59].
Workflow Integration for Multiplex ddPCR Applications
The integration of optimized pre-analytical procedures creates a standardized workflow that maximizes sample quality for multiplex ddPCR applications in ctDNA analysis. The following workflow diagram illustrates the complete pathway from blood collection to data analysis, highlighting critical decision points and quality control checkpoints.
Workflow for ctDNA Analysis in Multiplex ddPCR
For multiplex ddPCR applications specifically, pre-analytical considerations extend to ensuring sufficient cfDNA quantity and quality to support multiple parallel amplifications. The implementation of molecular barcoding strategies during library preparation can help reduce background noise and improve the sensitivity and specificity of mutation detection, which is particularly important when analyzing the low variant allele fractions characteristic of ctDNA [58]. Additionally, the use of locked nucleic acid (LNA) probes in ddPCR assays can enhance discrimination between wild-type and mutant alleles, further optimizing the analytical performance for ctDNA detection [16].
The reliable detection and quantification of ctDNA using multiplex ddPCR technologies is fundamentally dependent on rigorous management of pre-analytical variables. Standardized protocols for blood collection tube selection, centrifugation parameters, and sample storage conditions form an integrated system that preserves sample integrity from patient to analyzer. The implementation of preservative blood collection tubes, optimized two-step centrifugation, consistent -80°C storage, and comprehensive quality control measures collectively establish a robust foundation for accurate ctDNA analysis. As liquid biopsy applications continue to expand in cancer research and clinical management, adherence to these evidence-based pre-analytical practices will ensure the generation of reliable, reproducible data that can effectively inform therapeutic decisions and patient management strategies.
The analysis of circulating tumor DNA (ctDNA) presents a significant analytical challenge due to the need to detect single-nucleotide variants (SNVs) in a vast background of wild-type DNA. In the context of multiplex droplet digital PCR (ddPCR) for ctDNA analysis, achieving high specificity in allele discrimination is paramount for accurate cancer diagnosis, monitoring of minimal residual disease (MRD), and guiding targeted therapies. Specificity refers to the ability of an assay to perfectly distinguish the mutant target allele from the wild-type sequence, minimizing false-positive signals. Locked Nucleic Acids (LNA) are a class of nucleic acid analogs that, when incorporated into detection probes, significantly enhance this discriminatory power. This note details the application of LNA chemistry and other methodological considerations within multiplex ddPCR workflows to ensure high-fidelity allele discrimination for ctDNA analysis, a critical focus in modern cancer research and drug development.
The Chemistry of Locked Nucleic Acids (LNAs)
Structural Properties and Mechanism of Action
Locked Nucleic Acids (LNAs) are nucleic acid analogs characterized by a methylene bridge that connects the 2'-oxygen of the ribose ring to the 4'-carbon. This structural modification "locks" the sugar in the N-type (3'-endo) conformation, which is the preferred conformation for base stacking and hybridization. This pre-organization of the phosphate backbone reduces the entropic penalty upon binding to a complementary DNA or RNA target, leading to a dramatic increase in thermal stability (melting temperature, T~m~) of the duplex. A key property of LNA is that this increase in affinity is not uniform; the penalty for mismatched base pairing is significantly greater than for perfectly matched sequences. This differential effect is the foundation for its improved specificity. Fluorescence experiments using 2-aminopurine suggest that LNA modifications enhance base stacking in perfectly matched base pairs while simultaneously decreasing stabilizing stacking interactions in mismatched duplexes. Furthermore, studies have shown that LNAs do not alter the amount of counterions released upon duplex denaturation, indicating that the mechanism of enhanced stability and specificity is rooted in pre-organization and stacking effects rather than changes in electrostatic interactions [64].
Guidelines for LNA Probe Design for Mismatch Discrimination
The placement of LNA monomers within a probe is critical for optimizing mismatch discrimination. The beneficial effect on specificity is highly dependent on the sequence context, the identity of the mismatched base pair, and the modification pattern.
- Central Triplet Modification: A general and highly effective strategy is to incorporate a triplet of LNA residues centered on the position of the suspected mismatch (e.g., the SNP site). This configuration typically provides the largest discriminatory power (ΔmdT~m~) for most mismatch types [64].
- Exception for G–T Mismatches: A notable exception to the triplet rule involves G–T mismatches. In this case, discrimination can decrease when the guanine nucleotide at the mismatch site itself, or even the flanking nucleotides, are modified with LNA. Therefore, alternative designs should be explored for discriminating this specific mismatch [64].
- Chimeric Probes: For practical applications, LNA monomers are incorporated into DNA oligonucleotides to create chimeric LNA/DNA probes. This approach balances the enhanced stability and specificity of LNA with the cost-effectiveness and nuclease resistance of DNA. End-modified, gapped chimeric LNA/DNA oligomers have shown favorable properties for diagnostic applications [64].
Experimental Protocols for LNA-Based Allele Discrimination
Protocol: Designing LNA-Enhanced Probes for ddPCR
This protocol outlines the steps for designing and validating LNA-modified hydrolysis (TaqMan) probes for allele-specific discrimination in a ddPCR assay.
1. Probe and Primer Design: * Target Identification: Identify the sequence encompassing the SNV. The variant base should be positioned centrally within the probe sequence. * LNA Incorporation: Design two allele-specific probes: one for the wild-type and one for the mutant allele. Incorporate LNA monomers using the "triplet rule," placing three LNA residues with the central one directly at the SNP site. For example, for a SNP where the wild-type has an 'A' and the mutant has a 'G', the wild-type probe would have an LNA-modified 'A' at the central position. * Exception Handling: If the SNP involves a G-T mismatch, avoid modifying the G or its immediate flanking nucleotides. Instead, test designs where the LNA triplet is shifted or where only one or two LNA modifications are used. * Fluorophore Selection: Label the 5' end of each allele-specific probe with a different fluorophore (e.g., FAM and HEX/VIC). Ensure the quencher is a compatible non-fluorescent quencher (NFQ), typically at the 3' end. * Primer Design: Design PCR primers that amplify a short product (60-100 bp) suitable for ctDNA analysis. Ensure primers do not contain known polymorphisms and have similar melting temperatures. Verify specificity in silico.
2. Assay Optimization: * Annealing Temperature Gradient: Perform a ddPCR run with a temperature gradient around the predicted annealing temperature (e.g., 55°C to 65°C). The optimal temperature is the highest one that maintains robust, positive amplification for the perfect match while minimizing signal from the mismatch probe. * Probe and Primer Concentration Titration: Titrate probe and primer concentrations to maximize the separation between positive and negative droplets for each channel. A typical starting point is 900 nM for primers and 250 nM for probes.
3. Validation with Control Material: * Use synthetic oligonucleotides or cell line DNA with known genotype as controls. * Run the optimized duplex assay (both probes in the same reaction) to confirm no cross-talk between channels and specific detection of the intended allele.
Protocol: Multiplex ddPCR for Methylation-Based Cancer Detection
DNA methylation changes are early events in carcinogenesis. This protocol describes a multiplex ddPCR approach to detect tumor-specific methylation markers in ctDNA, which can be used for multi-cancer detection.
1. Sample Collection and Processing: * Blood Collection: Draw blood into cell-stabilizing blood collection tubes (e.g., cfDNA BCT by Streck) to prevent genomic DNA contamination from white blood cell lysis. Process within the prescribed time (e.g., up to 7 days at room temperature). * Plasma Separation: Centrifuge blood twice: first at 2,000 g for 10 minutes to isolate plasma, then at 12,000-20,000 g for 10 minutes at 4°C to remove residual cells and debris. * cfDNA Extraction: Extract cfDNA from plasma using a silica membrane-based kit (e.g., QIAamp Circulating Nucleic Acid Kit) or magnetic beads. Elute in a small volume (e.g., 60 µL) to maximize concentration [65] [6].
2. Bisulfite Conversion: * Treat extracted DNA (e.g., 20 ng) with bisulfite using a commercial kit (e.g., EZ DNA Methylation-Lightning Kit, Zymo Research). This process converts unmethylated cytosines to uracils, while methylated cytosines remain as cytosines. * Elute the converted DNA in a small volume (e.g., 15 µL) and use it promptly in the ddPCR reaction [6] [7].
3. Multiplex ddPCR Assay Setup: * Assay Design: Design primers and probes to target the bisulfite-converted sequence of differentially methylated regions (DMRs). Probes should distinguish between methylated (C-converted) and unmethylated (U-converted) alleles. The use of LNA in these probes can enhance discrimination. * Multiplexing: Combine assays for multiple DMRs in a single reaction, using a different fluorescent dye for each target. For example, a triplex assay for lung cancer detection might use FAM, HEX, and Cy5. * Droplet Generation and PCR: Combine the bisulfite-converted DNA sample with the ddPCR supermix, primers, and probes. Generate droplets using a droplet generator. Perform PCR amplification with a optimized thermal cycling protocol. * Droplet Reading and Analysis: Read the droplets on a droplet reader. Analyze the data to determine the concentration (copies/µL) of methylated and unmethylated alleles for each target. Apply a pre-defined cut-off to determine ctDNA-positivity [6] [7].
Performance Data and Applications
Quantitative Performance of LNA and ddPCR Assays
Table 1: Performance Metrics of LNA-Modified Probes and ddPCR Assays in Cancer Detection
| Assay Type / Chemistry | Application / Target | Key Performance Metric | Result / Value | Context / Notes |
|---|---|---|---|---|
| LNA-modified Probes [64] | Mismatch Discrimination | ΔmdT~m~ (Mismatch Discriminating ΔT~m~) | Largest for LNA triplets centered on mismatch | General rule; G-T mismatches are an exception. |
| Multiplex Methylation ddPCR [6] | Lung Cancer Detection (Metastatic) | ctDNA-Positive Rate | 70.2% - 83.0% | Varies with the statistical cut-off method used. |
| Multiplex Methylation ddPCR [6] | Lung Cancer Detection (Non-Metastatic) | ctDNA-Positive Rate | 38.7% - 46.8% | Highlights challenge of low tumor burden. |
| Triplex Methylation ddPCR [7] | Multi-Cancer Detection (8 types) | Cross-validated Area Under Curve (cvAUC) | 0.948 (94.8% accuracy) | Combined use of three targets improves performance over single targets. |
| MIL-based Multiplex-qPCR [66] | KRAS/BRAF SNP Detection | Enrichment Factor (vs. commercial kits) | >35 in buffer; superior in plasma | Magnetic Ionic Liquid (MIL) extraction improves preconcentration. |
Advanced Applications and Integrated Technologies
The combination of LNA with other sophisticated techniques further enhances detection capabilities. For instance, an electrochemical bioplatform for detecting the BRAF V600E mutation utilized LNA capture probes in conjunction with Rolling Circle Amplification (RCA). This dual detection system provided excellent selectivity for discriminating single-nucleotide variants without the need for PCR, demonstrating the versatility of LNA chemistry beyond optical detection methods [67]. Furthermore, the pre-analytical phase is critical. Methods such as using specialized blood collection tubes and optimizing centrifugation protocols are essential to preserve ctDNA and minimize background wild-type DNA, thereby improving the effective specificity and sensitivity of the final assay [65].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents and Materials for LNA-based Allele Discrimination Assays
| Item | Function / Application | Examples / Notes |
|---|---|---|
| LNA-Modified Probes | Allele-specific detection with enhanced specificity and T~m~. | Custom synthesized; design with central LNA triplet on SNP. |
| Cell-Stabilizing BCTs | Prevents release of wild-type gDNA from blood cells during storage/transport. | cfDNA BCT (Streck), PAXgene Blood ccfDNA (Qiagen). |
| Silica-Membrane cfDNA Kits | Efficient extraction of short-fragment cfDNA from plasma. | QIAamp Circulating Nucleic Acid Kit (Qiagen), Cobas ccfDNA Kit. |
| Bisulfite Conversion Kits | Converts unmethylated C to U for methylation analysis. | EZ DNA Methylation-Lightning Kit (Zymo Research). |
| ddPCR Supermix | Reagent mix for digital PCR with droplet stabilization. | ddPCR Supermix for Probes (Bio-Rad). |
| Magnetic Ionic Liquids (MILs) | Alternative DNA extraction solvent enabling preconcentration and direct integration into PCR. | [N₈,₈,₈,Bz⁺][Ni(hfacac)₃⁻]; used in multiplex-qPCR [66]. |
Workflow and Signaling Diagrams
Robust quality control (QC) is the cornerstone of reliable circulating tumor DNA (ctDNA) analysis, a critical component of liquid biopsy applications in precision oncology [27] [44]. The pre-analytical phase, particularly the extraction of cell-free DNA (cfDNA), profoundly impacts downstream analytical performance, including the sensitivity and specificity of multiplex droplet digital PCR (ddPCR) assays [68] [44]. ctDNA often represents less than 0.1% of total cfDNA in early-stage cancers, making its detection exceptionally vulnerable to suboptimal DNA yield, integrity, or the presence of contaminants [53] [27]. This application note provides detailed protocols and metrics for comprehensive QC assessment within the context of multiplex ddPCR-based ctDNA research, ensuring data integrity and supporting robust scientific conclusions.
Critical Quality Control Metrics and Assessment Methods
A tripartite QC strategy assessing extraction efficiency, DNA integrity, and contamination is essential for validating cfDNA samples. The following table summarizes the core metrics, their significance, and recommended assessment technologies.
Table 1: Essential Quality Control Metrics for ctDNA Analysis
| QC Category | Specific Metric | Significance in ctDNA Analysis | Recommended Assessment Method |
|---|---|---|---|
| Extraction Efficiency | Total cfDNA Yield | Impacts assay sensitivity; low yield limits mutant allele detection [44]. | Target-specific ddPCR [44] |
| Recovery of Spiked-in Control | Directly measures extraction kit performance and procedural efficacy [6]. | Spike-in DNA (e.g., CPP1) with ddPCR [6] | |
| DNA Integrity | Fragment Size Distribution | Confirms prevalence of mononucleosomal DNA (~167 bp); deviant sizes suggest gDNA contamination or degradation [44]. | Multiplex ddPCR sizing assays [44] or Capillary Electrophoresis |
| Short/Long Fragment Ratio | Higher ratios indicate enriched ctDNA; lower ratios suggest gDNA contamination [44]. | Multiplex ddPCR (e.g., OR gene family assay) [44] | |
| Contamination | Genomic DNA (gDNA) | gDNA contamination drastically dilutes ctDNA variant allele frequency (VAF) [44]. | ddPCR amplifying long genomic targets (>250 bp) [6] [44] |
| Lymphocyte DNA | Induces false negatives by diluting tumor-derived signals with wild-type DNA [6]. | Immunoglobulin gene-specific ddPCR assay (PBC) [6] |
Experimental Protocol: Assessing Extraction Efficiency and DNA Integrity via Multiplex ddPCR
The following protocol, adapted from Sánchez et al. (2020) [44], describes a multiplex ddPCR assay to simultaneously determine cfDNA concentration, fragment size distribution, and potential gDNA contamination in a single reaction.
Principle: The assay promiscuously cross-amplifies multiple targets within the human olfactory receptor (OR) gene family with amplicons designed for three different size ranges (e.g., 73-165 bp, 166-253 bp, and >253 bp). A separately probed, stable diploid reference locus (e.g., STAT6) provides absolute quantification for yield calculation [44].
Reagents and Equipment:
- QIAamp Circulating Nucleic Acid Kit (manual or QIAcube) [68]
- EZ DNA Methylation-Lightning Kit (for methylation studies) [6] [7]
- QIAsymphony SP with DSP Circulating DNA Kit (alternative) [6]
- Bio-Rad QX200 or QX600 Droplet Digital PCR System [53] [7]
- ddPCR EvaGreen or Probe Supermix
- Custom primers and probes for OR size fractions and reference gene
Procedure:
- cfDNA Extraction and Quantification: Extract cfDNA from 2-4 mL of plasma using a validated kit (e.g., QIAamp Circulating Nucleic Acid Kit). Elute in a minimal volume (e.g., 60 µL) [68].
- Reaction Setup: Prepare a 22 µL ddPCR reaction mix containing:
- 1X ddPCR Supermix
- Primers and probes for the three OR size fractions (e.g., FAM, HEX, Cy5 labels)
- Primers and probe for the reference gene (e.g., a distinct fluorescent label)
- 2-5 µL of extracted cfDNA (targeting 1-3 ng input based on preliminary quantification) [44]
- Droplet Generation and PCR: Generate droplets using the QX200/QX600 Droplet Generator. Perform PCR amplification with a standardized thermal cycling protocol.
- Droplet Reading and Analysis: Read the droplets on the QX200/QX600 Droplet Reader. Analyze the data using the associated software to obtain the absolute copy number for each target.
Data Analysis and Interpretation:
- Total cfDNA Yield (ng/µL): Calculate based on the copies/µL of the reference gene (STAT6), using the conversion factor that 1 ng of cfDNA ≈ 303 haploid genome equivalents [44].
- Fragment Size Distribution: Calculate the fractional abundance for each OR size fraction (Short, Medium, Long) as a percentage of the total OR-derived copies.
- gDNA Contamination Index: Compute the ratio of copies from the long fragment (>253 bp) to the short fragment (73-165 bp). A ratio exceeding a validated threshold (e.g., >0.1) indicates significant gDNA contamination [44].
- Assay Precision: Replicate testing (n≥2) is recommended. The ratio of short-to-medium-sized fragments should not vary by more than 10% between replicates [44].
Experimental Protocol: Evaluating Extraction Methods and Contamination
This protocol outlines a comparative approach for evaluating cfDNA extraction kits and assessing sample purity.
Principle: Different extraction methods exhibit varying efficiencies and recovery rates. This protocol compares multiple methods using healthy donor plasma spiked with a synthetic DNA control to directly quantify recovery and assess gDNA contamination [68].
Reagents and Equipment:
- Plasma samples from healthy donors (e.g., n=18) [68]
- Multiple cfDNA extraction kits (e.g., QIAamp Circulating Nucleic Acid Kit, QIAsymphony DSP Circulating DNA Kit) [68]
- Exogenous spike-in control (e.g., CPP1 DNA fragment) [6]
- ddPCR system and reagents for spike-in and gDNA targets
Procedure:
- Sample Preparation and Spike-in: Aliquot plasma from each healthy donor. Add a known quantity (~9000 copies/mL) of an exogenous spike-in DNA (CPP1) to each aliquot prior to extraction [6].
- Parallel Extraction: Extract cfDNA from the spiked plasma aliquots using different kits/methods according to manufacturers' instructions. Perform all extractions in a manner that minimizes day-to-day variability [68].
- Post-Extraction QC Analysis:
- Extraction Efficiency: Quantify the recovery of the spike-in CPP1 using a specific ddPCR assay. Higher recovery indicates better extraction efficiency [6].
- gDNA Contamination: Use a ddPCR assay amplifying a long genomic target (e.g., a 250 bp region of the EMC7 gene) to detect high-molecular-weight DNA contamination [6]. The ratio of a long (250 bp) to a short (65 bp) EMC7 amplicon can serve as a sensitive indicator [6].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Kits for ctDNA QC Workflows
| Item | Function/Application | Example Product/Catalog |
|---|---|---|
| cfDNA Extraction Kit | Manual, high-yield extraction from plasma/serum [68] | QIAamp Circulating Nucleic Acid Kit |
| Automated Extraction System | Automated, reproducible, high-throughput cfDNA extraction [6] | QIAsymphony SP with DSP Circulating DNA Kit |
| Bisulfite Conversion Kit | Conversion of unmethylated cytosines for methylation-specific ddPCR assays [6] [7] | EZ DNA Methylation-Lightning Kit |
| Exogenous Spike-in Control | Internal control for quantifying extraction efficiency and cfDNA recovery [6] | Synthetic CPP1 DNA Fragment |
| ddPCR System | Absolute quantification and rare allele detection for QC assays [6] [53] [44] | Bio-Rad QX200/QX600 Droplet Digital PCR System |
| gDNA Contamination Assay | Detects high-molecular-weight genomic DNA contamination [6] | EMC7 65/250 bp ddPCR Assay |
Workflow and Logical Pathway for Quality Control
The following diagram illustrates the sequential logic and decision points in a comprehensive QC workflow for ctDNA samples.
Quality Control Decision Workflow for ctDNA Analysis
Implementing rigorous, multi-faceted QC protocols is non-negotiable for generating reliable and reproducible data in multiplex ddPCR-based ctDNA research. The metrics and detailed protocols outlined herein—focusing on extraction efficiency with spike-in controls, DNA integrity via fragment sizing, and vigilant contamination monitoring—provide a foundational framework. Adherence to these standards ensures that pre-analytical variability is minimized, thereby safeguarding the analytical sensitivity required to detect low-frequency variants and empowering robust downstream clinical and research applications.
Validation Frameworks and Comparative Analysis with NGS and qPCR
In the field of molecular diagnostics using droplet digital PCR (ddPCR) for circulating tumor DNA (ctDNA) analysis, rigorous analytical validation is a critical prerequisite for generating reliable, clinically actionable data. The low abundance of ctDNA in patient plasma, which can sometimes constitute less than 0.01% of the total cell-free DNA, demands exceptionally sensitive and specific detection methods [9]. This application note details a standardized framework for determining three fundamental analytical performance indicators—Limit of Blank (LoB), Limit of Detection (LoD), and Precision—within the context of a research thesis focused on multiplex ddPCR for ctDNA analysis. These parameters are essential for validating the sensitivity, reliability, and robustness of ddPCR assays intended for cancer detection, prognosis, and longitudinal monitoring [9] [69]. The protocols herein are adapted from established clinical guidelines, such as the Clinical and Laboratory Standards Institute (CLSI) EP17-A2 standard, and tailored for ddPCR applications in ctDNA research [70].
Core Concepts and Definitions
- Limit of Blank (LoB): The LoB is defined as the highest apparent concentration of the target that is likely to be observed in a blank sample containing no target sequence. It is determined with a probability of
PLoB = 95%(where the false-positive rateα = 5%). In practice, it sets the false-positive cutoff, establishing an upper threshold for noise in the assay system [70]. - Limit of Detection (LoD): The LoD is the lowest concentration of the target that can be reliably detected in a sample. It is defined as the concentration at which the target can be distinguished from the LoB with a stated probability, typically
PLoD = 95%(where the false-negative rateβ = 5%). The LoD is a function of both the LoB and the variability observed in low-level positive samples [70]. - Precision: Precision refers to the closeness of agreement between independent measurement results obtained under stipulated conditions. In the context of ddPCR validation, it is assessed by measuring repeatability (within-run precision) and intermediate precision (across different days, operators, or instrument runs) using multiple replicates of samples with varying target concentrations [71] [72].
- Critical Sample Types:
- Blank Sample/Negative Control: A sample that does not contain the target sequence but is otherwise representative of the test sample matrix (e.g., wild-type plasma DNA for ctDNA assays) [70].
- Low-Level (LL) Sample: A positive sample with a target concentration near the expected LoD, typically between one and five times the LoB. This sample is used to measure the assay's variability at the detection limit [70].
Experimental Protocols
Protocol for Determining the Limit of Blank (LoB)
The following non-parametric method is recommended for calculating the LoB and requires a minimum of 30 independent blank sample replicates to achieve a 95% confidence level [70] [73].
- Sample Preparation and Analysis: Prepare and analyze a minimum of
N=30blank samples. These samples should mirror the biological matrix of the test samples (e.g., plasma DNA from a healthy donor for a ctDNA assay) and be processed identically through the entire ddPCR workflow, including nucleic acid extraction [70]. - Data Collection: For each target in the multiplex assay, export the measured concentration (in copies/µL) from the ddPCR software for all
Nblank replicates [70]. - Data Sorting: Sort the
Nconcentration values in ascending order (from Rank 1 to Rank N) [70]. - Rank Calculation: Calculate the rank position
Xcorresponding to the 95th percentile using the formula:X = 0.5 + (N * 0.95)For example, withN=30,X = 0.5 + (30 * 0.95) = 29[70]. - LoB Determination:
- If
Xis a whole number (e.g., 29.0), the LoB is the concentration value at that rank [70]. - If
Xis not a whole number (e.g., 29.4), identify the concentration values at the ranks immediately below (C1) and above (C2)X. The LoB is calculated by linear interpolation:LoB = C1 + Y*(C2 - C1), whereYis the decimal portion ofX[70].
- If
Table 1: Example LoB Calculation with N=30 Blank Samples
| Parameter | Value |
|---|---|
| Number of Blanks (N) | 30 |
| Target Percentile | 95% |
| Calculated Rank (X) | 29.0 |
| Concentration at Rank 29 | 0.05 copies/µL |
| Final LoB | 0.05 copies/µL |
Protocol for Determining the Limit of Detection (LoD)
The LoD calculation requires both the LoB and data from low-level (LL) positive samples to measure variability near the detection limit. This protocol uses a parametric approach, assuming the sample concentrations are normally distributed [70].
- Low-Level Sample Preparation: Prepare a minimum of five independently prepared LL samples. The target concentration should be within one to five times the previously determined LoB. These can be replicates of the same concentration or a narrow range of low concentrations [70].
- Replicate Analysis: For each of the
JLL samples, perform a minimum of six replicate ddPCR measurements. This results in a total of at least 30 data points (J=5,n=6) [70]. - Standard Deviation Calculation: Calculate the standard deviation (
SDi) for the replicate measurements of each LL sample. Test the homogeneity of variances between the LL samples using a statistical test like Cochran's test. If variances are not homogeneous, repeat the study with more appropriate LL samples [70]. - Pooled Standard Deviation: Calculate the global, pooled standard deviation (
SDL) across all LL samples using the formula:SDL = √( Σ[(ni - 1) * SDi²] / (L - J) )whereniis the number of replicates for theith LL sample,Jis the number of LL samples, andLis the total number of replicates. If all LL samples have the same number of replicates (n), the formula simplifies toSDL = √( ΣSDi² / J )[70]. - LoD Calculation:
Table 2: Example LoD Calculation Data
| Parameter | Value |
|---|---|
| LoB (from previous) | 0.05 copies/µL |
| Number of LL Samples (J) | 5 |
| Replicates per Sample (n) | 6 |
| Pooled SD (SDL) | 0.08 copies/µL |
| Cp Coefficient | ~1.645 |
| Calculated LoD | 0.18 copies/µL |
Protocol for Determining Precision
Precision is evaluated by analyzing multiple replicates of control samples at different concentrations (e.g., low, medium, high) across various experimental conditions.
- Sample Tiers: Define at least two concentration levels, including one near the LoD to assess precision at the detection limit [71] [72].
- Repeatability (Within-Run): Analyze a minimum of
n ≥ 5replicates of each control sample in a single run by the same operator using the same equipment and reagents. Calculate the mean, standard deviation (SD), and coefficient of variation (CV%) for each concentration level [72]. - Intermediate Precision (Between-Run): Analyze the same control samples across multiple different runs (e.g., different days, different operators, or different reagent batches). A minimum of
n ≥ 5replicates per level over at least 5 separate runs is recommended. Calculate the overall mean, SD, and CV% for the aggregated data [72]. - Data Analysis: The CV% is calculated as
(SD / Mean) * 100. Acceptable precision criteria are assay-dependent, but CVs below 25% are often targeted for low-concentration samples, with tighter thresholds (e.g., <10-15%) for higher concentrations [73].
Figure 1: Experimental Workflow for Analytical Validation. This diagram outlines the sequential process for determining LoB, LoD, and Precision.
The Scientist's Toolkit: Research Reagent Solutions
The following reagents and materials are essential for executing the validation protocols described above.
Table 3: Essential Reagents and Materials for ddPCR Validation
| Item | Function/Description | Example |
|---|---|---|
| Blank Sample Matrix | Provides the biological background for the assay without the target; critical for defining LoB. | Wild-type plasma/serum DNA for ctDNA assays [70]. |
| Low-Level Positive Control | Synthetic or cell-line DNA with known mutation used to prepare LL samples for LoD determination. | Linearized plasmid DNA or DNA from characterized mutant cell lines [74] [72]. |
| ddPCR Supermix | The master mix containing DNA polymerase, dNTPs, and buffer, optimized for droplet formation and stability. | Bio-Rad ddPCR Supermix for Probes [73]. |
| Mutation-Specific Assays | Primers and fluorescently labeled probes (e.g., TaqMan, PrimeTime) designed to specifically detect the target mutation(s). | Custom-designed TaqMan MGB or LNA-ZEN probes [74] [75]. |
| Droplet Generation Oil | Oil used to partition the PCR reaction into thousands of nanoliter-sized droplets. | DG-8 Cartridge Oil or Droplet Generation Oil for Probes [72]. |
| Cell-Free DNA Extraction Kit | For isolating high-quality, fragmented cfDNA from plasma samples. | QIAamp Circulating Nucleic Acid Kit, DSP Circulating DNA Kit [71] [6]. |
| Blood Collection Tubes (Streck/EDTA) | Tubes for blood draw that stabilize nucleated blood cells and prevent genomic DNA contamination of plasma. | Streck Cell-Free DNA BCT tubes or K2EDTA tubes [71]. |
Data Interpretation and Decision Framework
Once the LoB and LoD are established, they are used to interpret results from test samples. The following decision table provides a framework for classifying results based on the measured target concentration [70].
Table 4: Decision Framework for Sample Analysis Based on LoB and LoD
| Target Concentration [C] | Interpretation |
|---|---|
| C ≤ LoB | Not Detected. The result is not statistically different from the blank. |
| LoB < C < LoD | Detected but not Quantifiable. The target is present but the concentration cannot be reliably quantified with 95% confidence. |
| C ≥ LoD | Detected and Quantifiable. The target is present and can be reliably quantified. |
This framework is vital for accurately reporting low-level ctDNA findings, especially in contexts like minimal residual disease monitoring where ctDNA levels can be extremely low [71]. The precision data further informs researchers of the expected variability around the reported concentration, which is crucial for determining whether a change in ctDNA level over time is statistically significant [69].
The precise determination of LoB, LoD, and Precision is not merely a procedural formality but a foundational element of robust multiplex ddPCR assay development for ctDNA analysis. The protocols outlined here, grounded in international standards and current research practices, provide a clear path for researchers to validate their assays. By rigorously defining the limits of their detection systems and understanding the associated measurement variability, scientists can generate high-quality, reliable data. This, in turn, strengthens the translational potential of ddPCR-based liquid biopsies in cancer research, from early detection and prognosis to the longitudinal monitoring of treatment response.
The analysis of circulating tumor DNA (ctDNA) via liquid biopsy represents a transformative approach in oncology, offering a minimally invasive method for cancer detection, monitoring, and management. Multiplex droplet digital PCR (ddPCR) has emerged as a particularly powerful technique for ctDNA analysis, combining the absolute quantification and high sensitivity of digital PCR with the ability to simultaneously assess multiple biomarkers. This application note details the framework for the clinical validation of a multiplex ddPCR assay, focusing on establishing its analytical sensitivity, specificity, and clinical utility across well-defined patient cohorts. The data and protocols herein are framed within broader research on developing a robust multiplex ddPCR system for lung cancer detection, leveraging tumor-specific DNA methylation biomarkers to achieve high sensitivity and specificity across various disease stages [6].
The clinical performance of a multiplex ddPCR assay for ctDNA detection was evaluated across multiple patient cohorts, including healthy controls, patients with non-metastatic (stage I-III) disease, and patients with metastatic (stage IV) lung cancer. The assay incorporated five tumor-specific methylation markers, identified through in silico analysis of public methylation arrays, to maximize detection sensitivity [6].
Table 1: Performance of a Methylation-Specific Multiplex ddPCR Assay in Lung Cancer Detection
| Patient Cohort | Sensitivity (Cut-off Method 1) | Sensitivity (Cut-off Method 2) | Key Findings |
|---|---|---|---|
| Non-Metastatic (Stage I-III) | 38.7% | 46.8% | Demonstrates utility in early-stage disease where ctDNA levels are low. |
| Metastatic (Stage IV) | 70.2% | 83.0% | Significantly higher sensitivity reflects higher tumor burden. |
| Healthy Controls & Benign Disease | Specificity > 99% [7] | Specificity > 99% [7] | High specificity is critical for minimizing false positives. |
| Histological Subtypes | Higher sensitivities for Small Cell Lung Cancer and Squamous Cell Carcinoma. | Performance can vary by cancer type. |
The choice of statistical cut-off method for defining ctDNA positivity has a substantial impact on reported sensitivity and specificity, underscoring the need for careful optimization during assay validation [6]. Furthermore, the high specificity, consistently over 99% as demonstrated in a multi-cancer ddPCR study, is essential for ensuring clinical utility and avoiding unnecessary follow-up procedures in healthy individuals [7].
Detailed Experimental Protocols
Patient Cohort Selection and Sample Collection
Objective: To collect and process matched tissue and blood samples from well-characterized patient cohorts for assay development and validation.
Materials:
- Cohorts: Healthy controls, patients with benign lung diseases, and patients with non-metastatic and metastatic lung cancer [6].
- Sample Types: Formalin-Fixed Paraffin-Embedded (FFPE) tissue from primary tumors and normal lung; plasma from peripheral blood draws.
- Collection Tubes: 9 mL EDTA blood collection tubes.
- Protocol:
- Collect whole blood via venipuncture.
- Centrifuge EDTA tubes at 2,000 × g for 10 minutes within 4 hours of collection to separate plasma.
- Aliquot plasma and store immediately at -80°C to preserve cfDNA integrity [6].
Cell-free DNA Extraction and Bisulfite Conversion
Objective: To isolate high-quality cfDNA from plasma and convert it for methylation analysis.
Materials:
- Extraction Kit: DSP Circulating DNA Kit (Qiagen) on QIAsymphony SP [6].
- Bisulfite Conversion Kit: EZ DNA Methylation-Lightning Kit (Zymo Research) [6].
- Exogenous Control: ~9000 copies/mL of CPP1 spike-in DNA fragment to monitor extraction efficiency [6].
- Protocol:
- Thaw plasma at 5°C and centrifuge at 10,000 × g for 10 minutes.
- Add exogenous CPP1 control to plasma.
- Extract cfDNA from 4 mL plasma using the automated system, eluting in 60 µL buffer.
- Concentrate eluted DNA to 20 µL using an Amicon Ultra-0.5 Centrifugal Filter unit.
- Perform bisulfite conversion on concentrated DNA according to the kit instructions, eluting in 15 µL M-Elution Buffer [6].
Multiplex ddPCR Setup and Analysis
Objective: To detect and quantify methylated ctDNA targets using a multiplex ddPCR assay.
Materials:
- ddPCR System: QIAcuity series (Qiagen) or equivalent droplet-based system [76] [77].
- Assays: Primer and probe sets for five differentially methylated regions.
- Reagents: Anza 52 PvuII restriction enzyme (Thermo Scientific) to reduce background [76].
- Protocol:
- Prepare a 40 µL reaction mixture containing 10 µL of bisulfite-converted DNA, ddPCR master mix, target-specific primers and probes, and restriction enzyme.
- Generate approximately 20,000 droplets using a droplet generator.
- Perform PCR amplification with the following cycling conditions: 95°C for 5 min, then 45 cycles of 95°C for 15 s and 58-60°C for 1 min [6] [76].
- Read the plate on a droplet reader to measure fluorescence in each partition (FAM, HEX/VIC, etc.).
- Analyze data using instrument software (e.g., QIAcuity Software Suite) to calculate the absolute concentration (copies/µL) of each methylated target based on Poisson statistics [76] [77].
Diagram 1: Multiplex ddPCR ctDNA Analysis Workflow.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Multiplex ddPCR ctDNA Analysis
| Reagent / Solution | Function / Application | Example Products / Components |
|---|---|---|
| Nucleic Acid Extraction Kit | Isolation of high-quality cfDNA from plasma samples. | DSP Circulating DNA Kit (Qiagen) [6] |
| Bisulfite Conversion Kit | Converts unmethylated cytosine to uracil, enabling methylation-specific detection. | EZ DNA Methylation-Lightning Kit (Zymo Research) [6] [7] |
| ddPCR Master Mix | Optimized buffer, enzymes, and dNTPs for robust digital PCR amplification. | QIAcuity Probe PCR Kit (Qiagen) [76] |
| Methylation-Specific Assays | Primer and probe sets targeting differentially methylated CpG islands. | Custom-designed assays (e.g., for HOXA9 and other markers) [6] |
| Restriction Enzyme | Digests high-molecular-weight genomic DNA contaminations, improving assay specificity. | Anza 52 PvuII (Thermo Scientific) [76] |
| Exogenous Spike-in Control | Monitors efficiency of DNA extraction and bisulfite conversion. | CPP1 DNA fragment [6] |
Analytical and Clinical Validation Data
Beyond initial sensitivity/specificity measurements, full clinical validation requires assessing several key analytical parameters.
Table 3: Key Analytical Validation Parameters for a Multiplex ddPCR Assay
| Validation Parameter | Description | Typical Performance |
|---|---|---|
| Limit of Detection (LOD) | The lowest concentration of methylated target reliably detected. | Varies per marker; ddPCR is highly sensitive for low-abundance targets [76]. |
| Linearity & Dynamic Range | The range over which the measured concentration is linearly related to the true concentration. | High linearity (R² > 0.99) demonstrated in dPCR systems [76]. |
| Precision (Repeatability) | The agreement between replicate measurements. | Low intra-assay variability (e.g., median CV% of 4.5%) [76]. |
| Analytical Specificity | Ability to distinguish methylated from unmethylated alleles and avoid cross-reactivity. | Achieved via careful primer/probe design and use of restriction enzymes [76]. |
Longitudinal monitoring of ctDNA levels in patients undergoing treatment can reveal dynamic changes that correlate with treatment response and disease progression, offering potential for guiding therapy [6]. The high sensitivity of ddPCR makes it particularly suited for detecting minimal residual disease (MRD) and early relapse [50] [77].
Diagram 2: ddPCR Absolute Quantification Principle.
The clinical validation of a multiplex ddPCR assay for ctDNA analysis demonstrates a robust, cost-effective, and highly sensitive approach for lung cancer detection across disease stages. The data presented herein, derived from recent studies, confirm that such assays can achieve high specificity while providing clinically relevant sensitivities, particularly in metastatic disease. The outlined protocols provide a framework for researchers to validate their own assays. Future efforts should focus on validating these findings in larger, prospective cohorts and exploring the full potential of multiplex ddPCR in guiding treatment decisions and monitoring therapeutic response in real-time.
Droplet Digital PCR (ddPCR) has established itself as a powerful technology for the absolute quantification of nucleic acids, offering high sensitivity and precision without the need for standard curves [77]. In the context of circulating tumor DNA (ctDNA) analysis—a cornerstone of liquid biopsy applications for cancer management—the choice between singleplex and multiplex ddPCR assays carries significant implications for research efficiency, cost, and diagnostic capability [6] [38]. This application note provides a direct, data-driven comparison of these two approaches, framed within the broader thesis that multiplex ddPCR presents a robust, cost-effective solution for advancing ctDNA research, particularly when analyzing precious samples for multiple biomarkers simultaneously. We summarize quantitative performance data, provide detailed protocols for a representative multiplex experiment, and outline key reagent solutions to guide researchers and drug development professionals in optimizing their liquid biopsy workflows.
The following tables consolidate key performance metrics from recent studies, enabling a direct comparison of singleplex and multiplex ddPCR approaches.
Table 1: Comparative Analytical Performance of Singleplex vs. Multiplex ddPCR
| Performance Metric | Singleplex ddPCR | Multiplex ddPCR | Context and Notes |
|---|---|---|---|
| Limit of Detection (LoD) | Varies by target; can detect down to 0.5 copies/μL [78] | 1.4 to 2.9 copies/μL for different viral targets in a 9-plex assay [79] | LoD in multiplex remains excellent, with minimal compromise for most targets. |
| Sensitivity (Clinical) | 94.2% (HPV ctDNA detection) [80] | 90.6% (HPV ctDNA detection) [80] | Slight decrease in clinical sensitivity for the multiplex approach in a direct comparison. |
| Specificity (Clinical) | 98.6% (HPV ctDNA detection) [80] | 96.3% (HPV ctDNA detection) [80] | Slight decrease in clinical specificity for the multiplex approach. |
| Precision and Concordance | Used as a reference standard [79] | High concordance with singleplex; no statistically significant differences (Mann-Whitney test, p > 0.1) [79] | Multiplex results are highly reproducible and agree well with singleplex data. |
| Multiplexing Capacity | 1 target per reaction | Up to 9 targets demonstrated in a single reaction [79] | Higher-plex assays maximize information from limited samples. |
Table 2: Comparison of Workflow and Economic Factors
| Factor | Singleplex ddPCR | Multiplex ddPCR |
|---|---|---|
| Sample Volume Required | Higher total volume to analyze multiple targets | Lower sample consumption; multiple targets from a single reaction |
| Reagent and Consumable Cost | Higher cumulative cost for multiple reactions | Lower cost per data point; reagents and plates are shared across targets |
| Hands-on Time & Throughput | Lower throughput; more setup time for multiple wells | Higher throughput; streamlined setup for multi-target analysis |
| Data Complexity | Simple data analysis and interpretation | Requires advanced software for multi-channel fluorescence analysis |
| Assay Development | Relatively straightforward | Requires extensive optimization of primer/probe concentrations and conditions |
Experimental Protocol: A 9-Plex Viral Detection Assay
The following detailed methodology is adapted from a pioneering study that developed a one-step 9-plex RT-ddPCR assay for high-risk viruses, demonstrating the practical application of high-plex ddPCR in complex matrices [79].
Key Research Reagent Solutions
Table 3: Essential Materials for High-Plex ddPCR
| Item | Function | Example Product |
|---|---|---|
| ddPCR System | Partition generation, thermal cycling, and droplet fluorescence reading | QX600 Droplet Digital PCR System (Bio-Rad) [79] |
| One-Step RT-ddPCR Kit | Integrated reverse transcription and PCR amplification in a single mix | One-Step RT-ddPCR Advanced Kit for Probes (Bio-Rad) [79] |
| Primer/Probe Sets | Target-specific assays for amplification and detection | Custom-designed assays with FAM, HEX, ROX, Cy5, ATTO590 fluorophores [79] |
| Nucleic Acid Extraction Kit | Isolation of high-quality nucleic acids from complex samples | Enviro Wastewater TNA Kit (Promega) [79] |
Detailed Step-by-Step Procedure
Assay Design and Primer/Probe Preparation:
- Design primer and probe sets for all targets against conserved genomic regions. Use dual probes for a single target (e.g., SARS-CoV-2 N1 and N2) to reduce false negatives.
- Incorporate a combination of fluorophores (e.g., FAM, HEX, ROX, Cy5, ATTO590) with appropriate quenchers (e.g., ZEN/Iowa Black FQ).
- Split the primer/probe mixes into two groups based on their fluorescence signal intensity:
- ppmix A (High Targets): Final concentration of 900 nM primers/300 nM probes. (e.g., SARS-CoV-2 N1, Influenza A, Influenza B, Hepatitis A).
- ppmix B (Low Targets): Final concentration of 400-450 nM primers/100-150 nM probes. (e.g., RSV, Hepatitis E, External Control, SARS-CoV-2 N2, endogenous control B2M). This concentration difference creates separated clusters in the 2D plot.
Reaction Setup:
- Prepare the master mix on ice in a final volume of 20 μL:
- 5.0 μL One-Step RT-ddPCR Supermix
- 2.0 μL Reverse Transcriptase
- 1.0 μL of 300 mM Dithiothreitol (DTT)
- Primers and probes from ppmix A and ppmix B at their optimized final concentrations
- 5 μL of RNA template (or cfDNA for ctDNA applications)
- Nuclease-free H2O to volume
- Mix thoroughly by pipetting. Do not vortex.
- Prepare the master mix on ice in a final volume of 20 μL:
Droplet Generation:
- Transfer the entire 20 μL reaction mix to the sample well of a DG8 cartridge.
- Add 70 μL of droplet generation oil to the oil well.
- Place the cartridge and a rubber gasket in the droplet generator. The instrument will automatically generate approximately 20,000 nanoliter-sized droplets per sample.
Thermal Cycling:
- Carefully transfer the generated droplets from the cartridge to a semi-skirted 96-well PCR plate. Seal the plate with a foil heat seal.
- Place the plate in a thermal cycler and run the following protocol:
- Reverse Transcription: 50 °C for 60 minutes
- Enzyme Activation: 95 °C for 10 minutes
- Amplification (40 cycles):
- Denaturation: 94 °C for 30 seconds
- Annealing/Extension: 61 °C for 60 minutes (ramp rate of 2 °C/s)
- Enzyme Deactivation: 98 °C for 10 minutes
- Hold: 4 °C ∞
Droplet Reading and Data Analysis:
- Place the PCR plate in the QX600 Droplet Reader.
- Use the instrument's software (e.g., QuantaSoft) to read the fluorescence amplitude of each droplet in all available channels.
- The software will apply Poisson statistics to the fraction of positive and negative droplets to provide an absolute copy number (copies/μL) for each of the nine targets in the reaction.
- Exclude wells with fewer than 10,000 total droplets from the analysis.
Workflow Diagram
The following diagram illustrates the core procedural and decision-making workflow for a multiplex ddPCR experiment, from assay design to data interpretation.
The data and protocols presented herein strongly support the integration of multiplex ddPCR into ctDNA research workflows. The primary advantage of multiplexing is unparalleled efficiency: it conserves precious patient samples, reduces reagent costs, and significantly increases data output per run [79] [78]. While singleplex assays remain the gold standard for maximizing sensitivity for a single marker and may be preferable for applications requiring the ultimate limit of detection [80], the observed performance compromise in multiplex is often minimal and statistically insignificant for many targets [79].
For ctDNA research, where analyzing a panel of mutations or methylation markers is often necessary, multiplex ddPCR offers a compelling balance of performance and practicality. Its inherent tolerance to inhibitors—a common challenge in cfDNA samples—and its ability to provide absolute quantification without reference standards make it uniquely suited for liquid biopsy analysis [6] [77]. Furthermore, the technology's capability to detect subtle (less than two-fold) changes in target concentration with high precision is critical for monitoring treatment response or minimal residual disease (MRD) [81] [78].
In conclusion, when research demands comprehensive profiling from limited material, multiplex ddPCR emerges as a superior strategy. Its implementation accelerates discovery and development cycles, ultimately contributing to more personalized and effective cancer diagnostics and therapies.
The analysis of circulating tumor DNA (ctDNA) has emerged as a cornerstone of precision oncology, enabling non-invasive tumor profiling, monitoring of treatment response, and detection of minimal residual disease (MRD). Two primary technologies dominate this landscape: droplet digital PCR (ddPCR) and next-generation sequencing (NGS). For researchers and drug development professionals navigating this field, understanding the technical capabilities, limitations, and appropriate applications of each platform is paramount. This application note provides a structured comparison based on recent evidence, detailing protocols and performance metrics to guide experimental design and technology selection for ctDNA-based research and clinical development.
Performance Comparison: Sensitivity, Specificity, and Clinical Utility
Direct comparisons of ddPCR and NGS reveal a complex performance landscape where the optimal technology often depends on the specific clinical or research context.
Table 1: Direct Performance Comparison of ddPCR and NGS in Cancer Detection
| Cancer Type | Sample Type | ddPCR Sensitivity | NGS Sensitivity | Specificity | Key Findings | Citation |
|---|---|---|---|---|---|---|
| Localized Rectal Cancer | Pre-therapy plasma | 58.5% (24/41) | 36.6% (15/41) | Not specified | ddPCR detection was significantly higher (p=0.00075); associated with higher clinical tumor stage. | [23] [82] |
| HPV16-Oropharyngeal Cancer | Plasma | ~70% | ~70% | Not specified | Both technologies showed good and comparable sensitivity. | [83] |
| HPV16-Oropharyngeal Cancer | Oral Rinse | 8.3% | 75.0% | Not specified | NGS demonstrated superior sensitivity in oral rinse samples (p<0.001). | [83] |
| Lung Cancer | Plasma | Reference Standard | 98.5% | 98.9% | A specific NGS method (MAPs) showed high accuracy versus ddPCR down to 0.1% VAF. | [84] |
A study on localized rectal cancer provides a clear example of context-dependent performance. In pre-therapy plasma, a tumor-informed ddPCR assay detected ctDNA in 58.5% of patients, significantly outperforming a tumor-uninformed NGS panel which detected ctDNA in only 36.6% of the same patient cohort [23] [82]. This highlights ddPCR's potential advantage in targeted, tumor-informed applications.
In contrast, for detecting HPV16 DNA in oral rinse samples from patients with oropharyngeal cancer, NGS demonstrated a marked superiority with 75.0% sensitivity compared to just 8.3% for ddPCR [83]. This suggests that the optimal technology is heavily influenced by the sample matrix and the target biomarker.
The high accuracy of a specialized NGS approach using Molecular Amplification Pools (MAPs) for lung cancer cfDNA analysis demonstrates that advanced NGS methods can achieve performance on par with ddPCR, with the added benefit of broader genomic coverage [84].
Economic and Operational Considerations: Cost and Throughput Analysis
Beyond pure performance, practical considerations like cost, throughput, and operational complexity significantly influence technology selection in both research and clinical development.
Table 2: Economic and Operational Comparison of ddPCR and NGS
| Parameter | ddPCR | NGS | Notes |
|---|---|---|---|
| Cost per Test | ~$20 (SMA diagnosis) [85] | Higher | ddPCR operational costs reported 5–8.5-fold lower than NGS [23]. |
| Equipment & Reagent Costs | Lower initial investment | Significant capital and recurring costs | NGS requires substantial investment in sequencing instruments and reagents. |
| Multiplexing Capability | Limited (2-5 plex) [7] | High (50+ genes) [84] | New multiplex ddPCR assays for 8 cancers show promise [7]. |
| Workflow Complexity | Lower | Higher | NGS involves library prep, sequencing, and complex bioinformatics. |
| Turnaround Time | Shorter (hours) | Longer (days) | ddPCR provides rapid results for targeted analysis. |
Multiple studies highlight the significant cost advantage of ddPCR. In a detailed cost-analysis for spinal muscular atrophy diagnosis, the cost per test for ddPCR was approximately $20, compared to $70 for an alternative method [85]. In the context of ctDNA detection, researchers noted that the operational costs of ddPCR are 5–8.5-fold lower than those of NGS [23]. This cost-effectiveness makes ddPCR particularly attractive for focused, high-volume biomarker assays in both research and routine clinical monitoring.
While NGS has a higher per-test cost, its ability tointerrogate dozens to hundreds of genes simultaneously provides a vastly superior breadth of information. This makes NGS the preferred technology for comprehensive genomic profiling, especially when the genetic alterations are not known in advance [84]. The choice between the two technologies, therefore, often hinges on the specific requirement for breadth versus depth of genomic information.
Application Protocols: Detailed Experimental Methodologies
Protocol 1: Tumor-Informed ddPCR for ctDNA Detection in Rectal Cancer
This protocol is adapted from a study comparing ddPCR and NGS in localized rectal cancer [23] [82].
Step 1: Primary Tumor Sequencing
- Isolate DNA from formalin-fixed paraffin-embedded (FFPE) tumor tissue using a kit such as the QIAamp DNA Micro kit (Qiagen).
- Perform targeted NGS using a panel like the Ion AmpliSeq Cancer Hotspot Panel v2 (ThermoFisher) to identify somatic mutations.
- Select 1-2 mutations with the highest variant allele frequencies (VAF) for ddPCR assay design.
Step 2: Plasma Collection and cfDNA Isolation
- Collect patient blood in cell-free DNA blood collection tubes (e.g., Streck Cell Free DNA BCT).
- Centrifuge within 4 hours of venepuncture to separate plasma.
- Extract cfDNA from 4 mL plasma using a specialized kit (e.g., DSP Circulating DNA Kit on QIAsymphony SP).
- Elute DNA in a suitable buffer and quantify using a fluorometer (e.g., Qubit).
Step 3: Droplet Digital PCR
- Design custom ddPCR assays (FAM/HEX probes) for the selected mutations.
- Prepare reaction mix containing ddPCR Supermix, assays, and ~9 µL of extracted cfDNA.
- Generate droplets using a droplet generator (e.g., Bio-Rad QX200). A typical reaction partitions the sample into ~20,000 droplets.
- Perform PCR amplification on a thermal cycler.
- Read the plate using a droplet reader and analyze with vendor software (e.g., QuantaSoft).
- Allocate results as ctDNA-positive if any detectable ctDNA is present above a defined threshold (e.g., VAF ≥ 0.01%).
Protocol 2: Multiplex Methylation ddPCR for Multi-Cancer Detection
This protocol summarizes a novel approach for detecting eight cancer types using a methylation-based ddPCR multiplex [7].
Step 1: Marker Selection and Assay Design
- Identify differentially methylated regions (DMRs) through in-silico analysis of public methylation databases (e.g., TCGA).
- Select a panel of 3-5 markers that provide high cross-validated AUC (e.g., 0.948) across the target cancer types.
- Design methylation-specific PCR assays for the selected CpG sites.
Step 2: Sample Processing and Bisulfite Conversion
- Extract genomic DNA from tissue or cfDNA from plasma.
- Add an exogenous spike-in DNA fragment (e.g., CPP1) to plasma samples before extraction to monitor extraction efficiency.
- Perform bisulfite conversion on 20 ng of DNA using a commercial kit (e.g., EZ DNA Methylation-Lightning Kit, Zymo Research).
- Elute bisulfite-converted DNA in a small volume (e.g., 15 µL) and use within 10 days.
Step 3: Multiplex ddPCR Setup and Analysis
- Concentrate the converted DNA using a centrifugal filter unit (e.g., Amicon Ultra-0.5).
- Set up a multiplex ddPCR reaction using probes for the selected methylation markers.
- Include quality control assays: a spike-in recovery assay, an immunoglobulin gene assay to detect lymphocyte DNA contamination, and a reference gene assay (e.g., EMC7) to assess total cfDNA.
- Generate and amplify droplets as in Protocol 1.
- Analyze data and determine ctDNA status using a predefined cut-off method, comparing the methylation signal to limits of blank (LOB) and detection (LOD) established from healthy controls.
Research Reagent Solutions: Essential Materials for ctDNA Analysis
Table 3: Key Reagents and Kits for ddPCR and NGS Workflows
| Reagent Category | Product Example | Function in Workflow | Application Context |
|---|---|---|---|
| Blood Collection Tubes | Streck Cell Free DNA BCT | Preserves cfDNA by stabilizing blood cells | Pre-analytical; critical for all liquid biopsy studies |
| cfDNA Extraction Kits | QIAamp Circulating Nucleic Acid Kit; DSP Circulating DNA Kit | Isolves and purifies cfDNA from plasma | Pre-analytical; essential for all downstream analysis |
| ddPCR Supermix | ddPCR Supermix for Probes (Bio-Rad) | Enables PCR amplification in droplets | Core ddPCR reagent |
| Bisulfite Conversion Kits | EZ DNA Methylation-Lightning Kit (Zymo Research) | Converts unmethylated cytosines to uracils | Methylation-specific ddPCR or NGS |
| NGS Library Prep Kits | Ion AmpliSeq Library Kit 2.0 | Prepares cfDNA libraries for sequencing | Targeted NGS panels |
| Digital PCR Systems | QX200 Droplet Digital PCR (Bio-Rad); QIAcuity (Qiagen) | Partitions and amplifies samples for absolute quantification | Core ddPCR instrumentation |
The choice between ddPCR and NGS is not a matter of one technology being universally superior, but rather of matching the technology's strengths to the specific research or clinical question. ddPCR offers superior sensitivity and cost-effectiveness for monitoring known, predefined mutations, making it ideal for tracking minimal residual disease, assessing therapy response, and validating specific biomarkers in large cohorts. In contrast, NGS provides unparalleled breadth for discovering novel variants, comprehensive genomic profiling, and analyzing complex samples where the genetic landscape is not fully characterized.
Future developments in multiplex ddPCR, particularly using methylation biomarkers as demonstrated in the multi-cancer detection assay [7], promise to expand the application of this cost-effective technology. Meanwhile, advances in NGS error-correction methods, such as Molecular Amplification Pools [84], continue to push the sensitivity of sequencing closer to that of digital PCR. For a comprehensive research program, the most powerful strategy may often be a complementary one, using NGS for broad discovery and ddPCR for sensitive, longitudinal validation and monitoring of key biomarkers.
The evolution of polymerase chain reaction (PCR) technologies has fundamentally transformed molecular diagnostics and life science research. Following conventional PCR and quantitative real-time PCR (qPCR), digital PCR (dPCR) represents the third generation of this revolutionary technology [77]. Among dPCR platforms, droplet digital PCR (ddPCR) has emerged as a powerful tool for applications requiring precise nucleic acid quantification and rare variant detection [86]. This technical note provides a comprehensive comparison between ddPCR and qPCR, with specific focus on their respective capabilities for absolute quantification and detection of rare genetic variants, particularly within the context of circulating tumor DNA (ctDNA) analysis for cancer research.
The fundamental distinction between these technologies lies in their approach to quantification. While qPCR relies on relative quantification based on standard curves and amplification kinetics, ddPCR employs partitioning of samples into thousands of nanoliter-sized droplets, enabling absolute target quantification without calibration curves through Poisson statistical analysis [77]. This methodological difference confers significant advantages for ddPCR in detecting minute quantities of mutant DNA sequences against a background of wild-type molecules—a critical requirement for liquid biopsy applications in oncology [9].
Fundamental Technological Differences and Performance Comparison
Core Principles and Mechanisms
Quantitative PCR (qPCR) operates on the principle of monitoring amplification in real-time using fluorescent reporters. The cycle threshold (Ct), at which fluorescence crosses a predetermined threshold, is proportional to the starting quantity of the target nucleic acid [86]. Quantification requires comparison to standard curves generated from samples of known concentration, introducing potential variability and standardization challenges [86]. This relative quantification approach is highly effective for many applications but reaches limitations when precise absolute quantification or rare variant detection is required.
Droplet Digital PCR (ddPCR) fundamentally differs by partitioning each sample into thousands of individual nanoliter-sized water-in-oil droplets, effectively creating a separate PCR reaction in each partition [77]. Following endpoint amplification, each droplet is analyzed for fluorescence, categorizing them as positive (containing target sequence) or negative (no target sequence) [77]. The absolute concentration of the target molecule is then calculated using Poisson statistics based on the ratio of positive to negative droplets, eliminating the need for standard curves [86] [77].
Table 1: Core Technological Differences Between qPCR and ddPCR
| Parameter | Quantitative PCR (qPCR) | Droplet Digital PCR (ddPCR) |
|---|---|---|
| Quantification Method | Relative (based on standard curve) | Absolute (Poisson statistics) |
| Detection Principle | Real-time fluorescence during amplification | Endpoint fluorescence after amplification |
| Sample Partitioning | No partitioning (bulk reaction) | Partitioning into thousands of droplets |
| Sensitivity | Moderate (typically >0.1% mutant allele frequency) | High (can detect <0.01% mutant allele frequency) |
| Precision | Dependent on standard curve quality | High, especially at low target concentrations |
| Resistance to Inhibitors | Moderate | High (due to sample partitioning) |
| Throughput | High | Moderate to high |
| Cost per Reaction | Lower | Higher (especially for consumables) |
Analytical Performance Comparison
Recent comparative studies have quantitatively demonstrated the performance advantages of ddPCR for challenging applications. In ctDNA analysis for early-stage breast cancer, both ddPCR and plate-based digital PCR showed high sensitivity for detecting mutant alleles representing ≤ 0.1% of cell-free DNA, with >90% concordance in ctDNA positivity between platforms [53]. The exceptional sensitivity of ddPCR enables detection of rare mutants at frequencies as low as 0.001%-0.01% in ideal conditions, far surpassing the typical 1-5% detection limit of conventional qPCR [9].
For copy number variation analysis, ddPCR demonstrates superior precision compared to qPCR, particularly for targets with low abundance [87]. A comparative study of digital PCR platforms found that both droplet-based and nanoplate-based systems showed high precision across most analyses, with coefficients of variation (CV) typically below 10% for validated assays [87]. This precision is maintained even in the presence of PCR inhibitors, as partitioning effectively dilutes inhibitors across multiple reactions, reducing their impact on amplification efficiency [86].
Table 2: Quantitative Performance Comparison for ctDNA Detection
| Performance Metric | qPCR | ddPCR | Application Context |
|---|---|---|---|
| Limit of Detection (LoD) | ~1% mutant allele frequency | ~0.1% mutant allele frequency | Early-stage cancer detection [53] |
| Sensitivity | 78-85% (varies by application) | 89.2% in mCRC, 64.4% in localized tumors | Colorectal cancer detection [88] |
| Specificity | 82-95% (varies by application) | 96.7% | Lung cancer detection [6] |
| Precision (CV) | 10-25% at low concentrations | 5-15% at low concentrations | Rare variant quantification [87] |
| Dynamic Range | 5-7 logs | 4-5 logs (per run) | Nucleic acid quantification [86] |
Advantages of ddPCR in Absolute Quantification
The capacity for absolute quantification without standard curves represents one of ddPCR's most significant advantages [86] [77]. This capability eliminates potential variability introduced by standard curve preparation and interpolation, providing more reliable and reproducible results across experiments and laboratories [77]. In practice, this means researchers can directly quantify target molecules in units of copies per microliter without reference materials, streamlining workflows and reducing potential error sources.
The partitioning approach underlying ddPCR also enhances tolerance to PCR inhibitors, a common challenge in clinical samples [86]. By distributing inhibitors across thousands of partitions, their effective concentration in any single droplet is substantially reduced, maintaining amplification efficiency even in complex sample matrices [86]. This robustness is particularly valuable for ctDNA analysis, where sample quality and purity can vary considerably.
For gene expression studies and copy number variation analysis, ddPCR provides direct absolute quantification of target sequences, overcoming the normalization challenges associated with qPCR [87]. Studies comparing platform performance have demonstrated that ddPCR delivers highly precise and reproducible copy number measurements across different platforms, with high linearity (R² > 0.98) between expected and measured gene copies [87].
Advantages of ddPCR in Rare Variant Detection
The unparalleled sensitivity of ddPCR for detecting rare mutations amidst abundant wild-type sequences has established it as a cornerstone technology for liquid biopsy applications [9]. This capability stems from the massive sample partitioning that effectively enriches rare targets into individual droplets, where they can be amplified without competition from dominant sequences [77].
In oncology research, this rare variant detection capability has profound implications. Multiple studies have validated ddPCR for detecting and quantifying ctDNA in various cancer types, including lung [6], colorectal [88], breast [53], and pancreatic cancers [9]. In metastatic colorectal cancer, a methylation-specific ddPCR multiplex assay demonstrated 89.2% sensitivity for ctDNA detection, highlighting its utility for monitoring disease progression [88]. Similarly, in lung cancer, ddPCR-based approaches have enabled detection of tumor-specific methylation markers with high specificity (96.7%), facilitating non-invasive cancer detection and monitoring [6].
The clinical relevance of rare variant detection is further underscored by studies showing that ctDNA dynamics measured by ddPCR correlate strongly with treatment response and patient outcomes [88]. In metastatic colorectal cancer patients, ctDNA levels measured during treatment were significantly associated with progression-free survival (PFS) and overall survival (OS) [88]. Patients classified as good responders based on ctDNA dynamics showed median PFS and OS of 11.4 and 35.3 months, respectively, compared to 5.1 and 6.85 months for patients with progressive disease [88].
Diagram 1: Workflow comparison and key advantages of ddPCR versus qPCR. The ddPCR process enables absolute quantification and superior rare variant detection through massive sample partitioning and Poisson statistical analysis.
Application Notes: ddPCR for ctDNA Analysis in Cancer Research
Methylation-Specific ddPCR Multiplex Assay for Lung Cancer Detection
Background: Lung cancer management faces challenges in early detection and minimal residual disease monitoring. Circulating tumor DNA (ctDNA) analysis via liquid biopsy offers a non-invasive approach for cancer detection and monitoring [6].
Protocol:
- Sample Collection and Processing: Collect whole blood in EDTA tubes or specialized cell-free DNA blood collection tubes (e.g., cfDNA BCT from Streck). Process within 2-6 hours for EDTA tubes or within 7 days for stabilized tubes [65]. Perform double centrifugation: first at 2,000 × g for 10 minutes at room temperature, then at 12,000-20,000 × g for 10 minutes at 4°C to obtain platelet-free plasma [65].
- cfDNA Extraction: Extract cfDNA from 4 mL plasma using the DSP Circulating DNA Kit (Qiagen) on QIAsymphony SP according to manufacturer's instructions [6]. Elute in 60 μL elution buffer. Add approximately 9,000 copies/mL of exogenous spike-in DNA (CPP1) to monitor extraction efficiency [6].
- DNA Concentration and Bisulfite Conversion: Concentrate extracted DNA to 20 μL using Amicon Ultra-0.5 Centrifugal Filter units (Merck) [6]. Perform bisulfite conversion using EZ DNA Methylation-Lightning Kit (Zymo Research) according to manufacturer's protocol. Elute bisulfite-converted DNA in 15 μL M-Elution Buffer.
- ddPCR Reaction Setup: Prepare ddPCR reaction mix containing:
- 10 μL ddPCR Supermix for Probes (no dUTP)
- 1.8 μL of each primer (final concentration 900 nM)
- 0.5 μL of each probe (final concentration 250 nM)
- 5-8 μL bisulfite-converted DNA template
- Nuclease-free water to 20 μL total volume
- Droplet Generation: Transfer 20 μL reaction mix to DG8 Cartridge together with 70 μL Droplet Generation Oil for Probes. Generate droplets using QX200 Droplet Generator.
- PCR Amplification: Transfer droplets to 96-well PCR plate. Seal and perform amplification with following conditions:
- 95°C for 10 minutes (enzyme activation)
- 40 cycles of: 94°C for 30 seconds, 56°C for 60 seconds (annealing/extension)
- 98°C for 10 minutes (enzyme deactivation)
- 4°C hold
- Ramp rate: 2°C/second
- Droplet Reading and Analysis: Place plate in QX200 Droplet Reader. Analyze using QuantaSoft software with appropriate threshold settings. Calculate target concentration based on Poisson statistics.
Validation Parameters: Assess assay performance using the following metrics:
- Extraction Efficiency: Monitor using exogenous spike-in DNA (CPP1) [6]
- Specificity: Evaluate in healthy control samples (typically >95%) [6]
- Sensitivity: Determine in patient cohorts (reported 38.7-46.8% in non-metastatic, 70.2-83.0% in metastatic lung cancer) [6]
- Precision: Calculate coefficient of variation (%CV) between replicates (typically <10% for validated assays) [87]
MS-ddPCR Multiplex for Colorectal Cancer Monitoring
Background: Detection of minimal residual disease and early relapse remains challenging in colorectal cancer (CRC). ctDNA analysis offers potential for monitoring disease progression and treatment response [88].
Protocol:
- Sample Processing: Process plasma samples as described in Section 5.1. Ensure proper sample volume (minimum 4 mL plasma recommended) for adequate cfDNA yield.
- Multiplex Assay Design: Design primers and probes for multiple tumor-specific methylation markers. Include at least five markers to enhance detection sensitivity [88]. Select markers identified through bioinformatics analysis of methylation arrays (e.g., Illumina 450K arrays) [6].
- ddPCR Setup: Prepare reaction mix similar to Section 5.1, with optimization for multiple probe channels. Use different fluorescent dyes (FAM, HEX/VIC, etc.) for distinct targets.
- Droplet Generation and Amplification: Follow same procedure as Section 5.1 steps 5-6.
- Data Analysis: Use multi-channel detection in QuantaSoft. Apply appropriate thresholds for each fluorescent channel. Calculate mutant allele frequency for each marker.
Performance Characteristics: A validated MS-ddPCR multiplex for CRC demonstrated:
- Specificity: 96.7% in control samples [88]
- Sensitivity: 64.4% in localized tumors, 89.2% in metastatic CRC [88]
- Prognostic Value: Significant association between ctDNA dynamics and progression-free survival (PFS) and overall survival (OS) [88]
Research Reagent Solutions
Table 3: Essential Reagents and Materials for ddPCR-based ctDNA Analysis
| Category | Specific Product | Manufacturer | Application Notes |
|---|---|---|---|
| Blood Collection Tubes | cfDNA BCT Tubes | Streck | Enable blood sample stability for up to 7 days at room temperature [65] |
| PAXgene Blood ccfDNA Tubes | Qiagen | Preserve cfDNA quality for 3-7 days at 4-25°C [65] | |
| Nucleic Acid Extraction | DSP Circulating DNA Kit | Qiagen | Optimized for cfDNA extraction from plasma samples [6] |
| QIAamp Circulating Nucleic Acid Kit | Qiagen | Silica-membrane based extraction with high recovery [65] | |
| Bisulfite Conversion | EZ DNA Methylation-Lightning Kit | Zymo Research | Rapid bisulfite conversion for methylation analysis [6] |
| ddPCR Master Mix | ddPCR Supermix for Probes | Bio-Rad | Standard reaction mix for probe-based detection |
| Droplet Generation | Droplet Generation Oil | Bio-Rad | Specialized oil for stable water-in-oil emulsion |
| DG8 Cartridges | Bio-Rad | Consumables for droplet generation | |
| Quality Control | Exogenous Spike-in Controls | Synthetic DNA fragments | Monitor extraction efficiency and PCR inhibition [6] |
The comparative analysis between ddPCR and qPCR reveals distinct advantages of ddPCR technology for applications requiring absolute quantification and rare variant detection. The capacity for precise, standard-free quantification combined with exceptional sensitivity for low-abundance targets positions ddPCR as a transformative technology in molecular diagnostics, particularly for liquid biopsy applications in oncology.
The proven utility of ddPCR in detecting ctDNA for lung [6], colorectal [88], breast [53], and pancreatic cancers [9], along with its demonstrated prognostic value for monitoring treatment response [88], underscores its growing importance in cancer research and clinical applications. While qPCR remains a robust, cost-effective solution for many molecular detection needs, ddPCR provides critical advantages for the most challenging detection scenarios where maximum sensitivity and precise quantification are paramount.
As liquid biopsy technologies continue to evolve, ddPCR is poised to play an increasingly central role in cancer detection, monitoring, and personalized treatment selection. Its unique capabilities address fundamental challenges in ctDNA analysis, making it an indispensable tool in the advancing field of molecular oncology.
Conclusion
Multiplex ddPCR has firmly established itself as a powerful, precise, and cost-effective tool for ctDNA analysis, playing a critical role in advancing cancer research and drug development. Its unparalleled sensitivity for detecting rare variants and absolute quantification capabilities make it indispensable for applications ranging from early cancer detection and MRD monitoring to real-time therapy guidance. Future directions will be shaped by technological integration, including the use of AI for assay optimization and data analysis, the development of higher-plexing capabilities, and the creation of portable, point-of-care systems. The ongoing standardization of protocols and the accumulation of robust clinical validation data will be paramount for its transition from a research tool to a fully integrated component of routine clinical oncology, ultimately enabling more personalized and effective cancer management.
References
- 1. Circulating tumor nucleic acids: biology, release mechanisms ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-022-01710-w]
- 2. Circulating tumor DNA: a promising biomarker in the liquid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5217053/]
- 3. Using cfDNA and ctDNA as Oncologic Markers [https://pmc.ncbi.nlm.nih.gov/articles/PMC10487653/]
- 4. Circulating tumor DNA to monitor treatment response in ... [https://www.nature.com/articles/s41698-025-00876-y]
- 5. Liquid biopsy in cancer: current status, challenges and ... [https://www.nature.com/articles/s41392-024-02021-w]
- 6. Development and validation of a methylation-specific ... [https://www.nature.com/articles/s41598-025-15228-w]
- 7. Simultaneous detection of eight cancer types using a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11705734/]
- 8. A Review of Circulating Tumor DNA (ctDNA) and the Liquid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12032849/]
- 9. Circulating Tumor DNA Detection by Digital-Droplet PCR in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7956845/]
- 10. dPCR: A Technology Review - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC5948698/]
- 11. dPCR: A Technology Review [https://www.mdpi.com/1424-8220/18/4/1271]
- 12. Droplet digital PCR as alternative to microbiological culture ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10925636/]
- 13. Multiplexed ddPCR-amplicon sequencing reveals isolated ... [https://www.nature.com/articles/s41467-023-39417-1]
- 14. Optimization of digital droplet polymerase chain reaction ... [https://www.sciencedirect.com/science/article/pii/S2214753515300127]
- 15. Comparing single‐target and multitarget approaches for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9580876/]
- 16. Optimisation of robust singleplex and multiplex droplet ... [https://www.nature.com/articles/s41598-019-49043-x]
- 17. ddpcr: an R package and web application for analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5031129/]
- 18. Development and evaluation of a multiplex droplet digital ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.970973/full]
- 19. Optimization of digital droplet polymerase chain reaction for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4827695/]
- 20. Digital droplet PCR is an accurate and precise method to ... [https://www.nature.com/articles/s41598-025-20944-4]
- 21. Direct comparison study of DNA methylation markers in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5641111/]
- 22. A comparative study of four cell-free DNA assays for detecting ... [https://breast-cancer-research.biomedcentral.com/articles/10.1186/s13058-025-02077-8]
- 23. Performance Comparison of Droplet Digital PCR and Next ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12062871/]
- 24. Droplet digital PCR for detection and quantification of ... [https://bmccancer.biomedcentral.com/articles/10.1186/s12885-017-3424-0]
- 25. Optimized (Pre) Analytical Conditions and Workflow for ... [https://www.sciencedirect.com/science/article/pii/S1525157819300169]
- 26. ctDNA detected by ddPCR reveals changes in tumour load ... [https://www.nature.com/articles/s41598-019-53917-5]
- 27. Monitoring and Assessment of Circulating Tumor DNA in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550271/]
- 28. Up-front mutation detection in circulating tumor DNA by ... [https://www.sciencedirect.com/science/article/pii/S1936523322002480]
- 29. Increased blood draws for ultrasensitive ctDNA and CTCs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11096188/]
- 30. Clinical Validation of Digital PCR-based ctDNA detection ... [https://www.medrxiv.org/content/10.1101/2025.07.02.25330747v1]
- 31. Computational tradeoffs in multiplex PCR assay design for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1190169/]
- 32. Design, validation, and implementation of multiplex digital ... [https://pubmed.ncbi.nlm.nih.gov/39701810]
- 33. Clinical validation of droplet digital PCR assays in ... [https://pubmed.ncbi.nlm.nih.gov/40250457/]
- 34. Evaluation of commercial kits for isolation and bisulfite ... [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-023-01563-0]
- 35. An automated high throughput solution for DNA extraction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6716871/]
- 36. The impact of multicancer early detection tests on cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12598375/]
- 37. Transforming Early Cancer Detection With the ... [https://www.healthcareexecutive.org/sponsored-content/transforming-early-cancer-detection-with-the-cancerguard-test]
- 38. Clinical applications of cell-free DNA-based liquid biopsy ... [https://www.sciencedirect.com/science/article/pii/S1936523325002505]
- 39. Multi-cancer early detection via a DNA methylation ... [https://pubmed.ncbi.nlm.nih.gov/40317486/]
- 40. Droplet Digital PCR Assay for Detection and Monitoring of ... [https://pubmed.ncbi.nlm.nih.gov/40627526/]
- 41. Whole-genome informed circulating tumor DNA analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10272573/]
- 42. Digital PCR: from early developments to its future application ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12278255/]
- 43. Three-color crystal digital PCR [https://www.sciencedirect.com/science/article/pii/S2214753516300328]
- 44. Evaluating the quantity, quality and size distribution of cell ... [https://www.nature.com/articles/s41598-020-69432-x]
- 45. Comparison of digital PCR systems for the analysis ... [https://www.sciencedirect.com/science/article/pii/S0009898123000414]
- 46. A clinician's handbook for using ctDNA throughout the patient ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-022-01551-7]
- 47. Understanding BLOODPAC's Analytical Validation Protocols [https://www.bloodpac.org/bloodpac-blog/understanding-bloodpacs-analytical-validation-protocols-for-ctdna-assays]
- 48. Generic Protocols for the Analytical Validation of Next ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7462123/]
- 49. Tracking Cancer Evolution through the Disease Course [https://pmc.ncbi.nlm.nih.gov/articles/PMC7611362/]
- 50. Real-World Technical Hurdles of ctDNA NGS Analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12564474/]
- 51. What's The Key To ctDNA Detection? Good Preparation [https://www.twistbioscience.com/blog/science/cfDNA-library-preparation-kit]
- 52. Guide to multiplexing dPCR assays [https://www.qiagen.com/us/applications/digital-pcr/beginners/dpcr-multiplexing]
- 53. Comparative study of droplet-digital PCR and absolute Q ... [https://www.sciencedirect.com/science/article/pii/S0009898123004758]
- 54. Guidelines for PCR Optimization with Thermophilic DNA ... [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/guidelines-for-pcr-optimization-with-thermophilic-dna-polymerases?srsltid=AfmBOoql_Y2SSFM1SEzcygJkqJ4CvDK8kAemeSw9MrWn4RgMCAyx0lME]
- 55. Polymerase Chain Reaction: Basic Protocol Plus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4846334/]
- 56. Stringent Base Specific and Optimization-Free Multiplex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8616434/]
- 57. PCR Cycling Parameters—Six Key Considerations for ... [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-cycling-considerations.html]
- 58. ctDNA Sequencing Services [https://www.cd-genomics.com/ctdna-sequencing-services.html]
- 59. Optimization of Sources of Circulating Cell-Free DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8647427/]
- 60. The impact of preanalytical variables on the analysis of cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11111958/]
- 61. or Vacutainer: Colors, Blood , Uses & Order of Draw Tube Types [https://myhematology.com/lab-protocols/blood-collection-tube-types/]
- 62. Vacutainer - Wikipedia [https://en.wikipedia.org/wiki/Vacutainer]
- 63. : How to Pick the Correct One Blood Collection Tubes [https://bitesizebio.com/23701/choosing-the-right-blood-collection-tubes/]
- 64. Design of LNA probes that improve mismatch discrimination [https://pmc.ncbi.nlm.nih.gov/articles/PMC1456327/]
- 65. Improvement of the sensitivity of circulating tumor DNA-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12332530/]
- 66. Allelic discrimination between circulating tumor DNA ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267020305110]
- 67. Dual detection system for cancer-associated point ... [https://www.sciencedirect.com/science/article/pii/S0925400523010900]
- 68. Extraction of Cell-Free DNA: Evaluation of Efficiency, ... [https://www.sciencedirect.com/science/article/pii/S152515782400014X]
- 69. Single-Color Digital PCR Provides High-Performance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6593258/]
- 70. How to characterize the Limit of Blank and ... [https://www.stillatechnologies.com/digital-pcr/naica-system-analysis/how-to-characterize-the-limit-of-blank-and-the-limit-of-detection-in-crystal-digital-pcr/]
- 71. Quantitative ctDNA Detection in Hepatoblastoma [https://pmc.ncbi.nlm.nih.gov/articles/PMC10778269/]
- 72. Evaluation of droplet digital PCR and next generation ... [https://www.nature.com/articles/s41598-018-27368-3]
- 73. Development and validation of a droplet digital PCR assay ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1573949/full]
- 74. Determining lower limits of detection of digital PCR assays ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5129438/]
- 75. A Novel Multiplex Droplet Digital PCR Assay to Identify and ... [https://www.sciencedirect.com/science/article/pii/S1525157818301144]
- 76. Digital PCR outperforms quantitative real-time PCR for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12288185/]
- 77. Digital PCR: from early developments to its future application ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00055f]
- 78. qPCR vs. ddPCR: Which Is Best for You? [https://www.bioradiations.com/qpcr-vs-ddpcr-assays-select-the-best-gene-expression-analysis-method-for-your-application1125/]
- 79. One Assay, Nine Targets: Advancing Viral Surveillance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12529474/]
- 80. Direct Comparison of Alternative Blood-Based Approaches for ... [https://pubmed.ncbi.nlm.nih.gov/40392113/]
- 81. Multiplex ddPCR Offers Superior Sensitivity Over qPCR [https://www.technologynetworks.com/analysis/blog/multiplex-ddpcr-offers-new-precision-for-gene-expression-analysis-406631]
- 82. Performance Comparison of Droplet Digital PCR and Next- ... [https://pubmed.ncbi.nlm.nih.gov/40346007/]
- 83. Comparison of next generation sequencing, droplet digital ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9058207/]
- 84. Sensitivity, specificity, and accuracy of a liquid biopsy ... [https://www.nature.com/articles/s41598-021-89592-8]
- 85. Cost estimation for spinal muscular atrophy diagnosis ... [https://ruralneuropractice.com/cost-estimation-for-spinal-muscular-atrophy-diagnosis-using-multiplex-ligation-dependent-probe-amplification-and-droplet-digital-polymerase-chain-reaction/]
- 86. Quantitative or digital PCR? A comparative analysis for ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993624001584]
- 87. Comparing the precision of two digital PCR applications for ... [https://www.nature.com/articles/s41598-025-13143-8]
- 88. Detection of circulating tumor DNA in colorectal cancer ... [https://pubmed.ncbi.nlm.nih.gov/41239188/]