xCell 2.0: A Comprehensive Guide to Tumor Microenvironment Deconvolution for Cancer Research and Therapy Development
This article provides a detailed exploration of the xCell algorithm and its advanced version, xCell 2.0, for digital dissection of the tumor microenvironment (TME) from bulk transcriptomics data.
xCell 2.0: A Comprehensive Guide to Tumor Microenvironment Deconvolution for Cancer Research and Therapy Development
Abstract
This article provides a detailed exploration of the xCell algorithm and its advanced version, xCell 2.0, for digital dissection of the tumor microenvironment (TME) from bulk transcriptomics data. Aimed at researchers, scientists, and drug development professionals, we cover the foundational principles of cell type enrichment analysis, methodological guidance for application using custom and pre-trained references, optimization strategies for robust results, and comprehensive validation against established benchmarks. The content synthesizes recent advancements demonstrating xCell 2.0's superior performance in predicting immunotherapy response and clinical outcomes, offering practical insights for leveraging this powerful tool in precision oncology and therapeutic development.
Decoding Cellular Heterogeneity: Foundational Principles of xCell TME Analysis
The Critical Role of TME Deconvolution in Precision Oncology
The tumor microenvironment (TME) is a dynamic ecosystem consisting of various cell types and processes that play a crucial role in tumor initiation, growth, progression, metastasis, and response to therapy [1] [2]. A detailed characterization of the TME and its association with genomic and clinical features is essential for deepening our understanding of tumor biology and resistance mechanisms. While single-cell transcriptomics represents the gold standard for TME analysis, this approach faces limitations including potential loss of cell types during sample preparation and high costs [1]. Computational deconvolution methods meet this need by inferring the relative proportions of specific cell types from bulk RNA-seq or microarray transcriptional profiles, enabling researchers to extract valuable TME information from existing large-scale databases such as The Cancer Genome Atlas (TCGA) [3].
The xCell algorithm represents a significant advancement in this field, providing a gene signature-based method that estimates the relative abundance of different cell types in bulk gene expression data [4] [5]. The recently introduced xCell 2.0 features an improved methodology including automated handling of cell type dependencies and more robust signature generation, allowing researchers to utilize any reference dataset for deconvolution analysis [4]. This algorithm has demonstrated superior performance in benchmarking studies, showing the best performance in minimizing spillover effects between related cell types and significantly improving prediction accuracy for immune checkpoint blockade response compared to other methods [4].
Key Methodological Advances in xCell 2.0
Technical Improvements and Algorithmic Enhancements
xCell 2.0 introduces a pipeline for generating custom reference objects that can be used for cell type enrichment analysis, significantly enhancing the method's applicability to diverse tissue types and experimental conditions [4]. Key improvements include:
Ontological Integration: xCell 2.0 automates the identification of lineage relationships among cell types using ontology IDs extracted directly from the standardized Cell Ontology (CL), enabling the entire pipeline to account for cell type dependencies automatically [4]. This addresses a critical limitation in deconvolution methods that require manual intervention to avoid lineage-related biases.
Enhanced Signature Generation: The algorithm generates cell type signatures using an improved approach that modifies the threshold criteria for determining gene inclusion. Instead of comparing against the top three other cell types, xCell 2.0 implements a threshold-based approach of at least 50% of the cell types in the reference, making it more adaptable to custom references with variable numbers of cell types [4].
Spillover Correction: xCell 2.0 uses in-silico simulated cell type mixtures to learn parameters that model the linear relationship between signatures' enrichment scores and cell type proportions. The method includes a spillover correction strength (α) parameter that allows users to balance between correcting for genuine spillover effects and potentially over-correcting [4].
Performance Validation and Benchmarking
In comprehensive benchmarking against eleven popular deconvolution methods using nine human and mouse reference sets and 26 validation datasets encompassing 1711 samples and 67 cell types, xCell 2.0 demonstrated superior accuracy and consistency across diverse biological contexts [4]. The algorithm also showed the best performance in the independent Deconvolution DREAM Challenge dataset, establishing its robustness for TME analysis [4].
The DREAM Challenge assessment, which evaluated six published and 22 community-contributed methods using in vitro and in silico transcriptional profiles of admixed cancer and healthy immune cells, revealed that while most methods predict coarse-grained populations well, several methods including xCell showed improved prediction of fine-grained populations [3]. This challenge also demonstrated the applicability of deep learning to deconvolution as an alternative methodology to previously employed reference- and enrichment-based approaches [3].
Experimental Protocols for TME Deconvolution Using xCell
Protocol 1: Pan-Cancer TME Profiling and Survival Association Analysis
Objective: To characterize TME composition across multiple cancer types and associate specific immune patterns with clinical outcomes using xCell.
Materials:
- Bulk RNA-seq data from tumor samples (e.g., TCGA dataset)
- xCell 2.0 algorithm (available as Bioconductor package or web application)
- Clinical annotation data including survival information
- Statistical analysis software (R recommended)
Methodology:
- Data Preparation: Obtain bulk gene expression data from tumor samples and corresponding clinical metadata. Ensure proper normalization and quality control.
- xCell Analysis: Apply xCell 2.0 to estimate abundances of 64 immune and stromal cell types. Use pre-trained references appropriate for your tissue type or generate custom references if necessary.
- Data Integration: Combine xCell output with clinical data, ensuring sample identifiers are properly matched.
- Stratification: Classify samples into TME clusters based on immune cell infiltration patterns using unsupervised clustering methods.
- Survival Analysis: Perform Kaplan-Meier analysis and Cox proportional hazards regression to assess the association between TME clusters and patient survival outcomes.
- Statistical Validation: Apply multiple testing correction and validate findings in independent cohorts when possible.
Expected Outcomes: Identification of distinct TME clusters with significant associations to patient survival. For example, application of this protocol revealed that leukocyte abundance showed negative correlation with risk of progression pan-cancer (hazard ratio HRadj = 0.73, p = 2.15e-06, n = 6406), with immune-rich TME clusters predicting better survival in specific cancer subtypes [1].
Protocol 2: Predictive Modeling of Therapy Response
Objective: To develop models predicting response to cancer therapy based on TME composition deconvolved using xCell.
Materials:
- Bulk RNA-seq data from pre-treatment tumor samples
- Treatment response data (e.g., pathological complete response vs. non-response)
- xCell 2.0 algorithm
- Machine learning libraries (e.g., scikit-learn, caret)
Methodology:
- TME Deconvolution: Process bulk gene expression data using xCell 2.0 to obtain immune and stromal cell scores.
- Feature Selection: Identify the most informative cell types for response prediction using univariate analysis or feature importance measures.
- Model Training: Develop predictive models using machine learning algorithms (e.g., random forest, logistic regression) with xCell-derived features as predictors.
- Model Validation: Assess model performance using cross-validation and independent validation cohorts.
- Comparison to Alternatives: Compare predictive performance against models using only cancer type and treatment information or bulk expression features.
Expected Outcomes: Development of robust predictors of therapy response. In a pan-cancer immune checkpoint blockade response prediction example, xCell 2.0-derived TME features significantly improved prediction accuracy compared to models using only cancer type and treatment information, and outperformed other deconvolution methods and established prediction scores [4].
Comparative Performance Analysis of Deconvolution Methods
Benchmarking Results Across Multiple Studies
Table 1: Performance Comparison of Major Deconvolution Methods
| Method | Type | Key Strengths | Limitations | Reported Performance |
|---|---|---|---|---|
| xCell 2.0 | Signature-based | Superior accuracy and consistency, minimal spillover effects, best performance in ICB response prediction | Cannot compare different cell types directly | Outperformed 11 other methods across 26 validation datasets [4] |
| Bisque | Reference-based | Accurate for brain tissue, effective assay bias correction | Variable performance across tissues | Most accurate for brain tissue in multi-assay benchmark [6] |
| hspe (dtangle) | Reference-based | Good performance in brain tissue, handles high collinearity | Limited benchmarking in cancer | Among top performers in brain tissue benchmark [6] |
| CIBERSORTx | Machine learning | Broadly used, good for coarse-grained cell types | Lower performance on fine-grained populations | Robust for coarse-grained but not fine-grained populations [3] [6] |
| DWLS | Reference-based | Weighted least squares approach | Variable performance across tissues | Moderate performance in brain tissue benchmark [6] |
| MuSiC | Reference-based | Source bias correction | Inconsistent performance across benchmarks | Variable performance in independent assessments [6] |
| BayesPrism | Bayesian | Bayesian framework | Computational intensity | Not top performer in brain benchmark [6] |
Integrated Approaches for Enhanced Deconvolution
Recent research has demonstrated that integrating multiple deconvolution tools can provide more comprehensive TME analysis than any single method. One pan-cancer study integrated nine deconvolution tools to assess 79 TME cell types in 10,592 tumors across 33 different cancer types, creating integrated scores (iScores) that showed improved correlation with ground truth measurements compared to individual tools [1] [2]. This integrated approach identified 41 patterns of immune infiltration and stroma profiles, revealing unique TME portraits for each cancer type and identifying a shared immune-rich TME cluster that predicts better survival in specific cancer subtypes [1].
The iScore approach demonstrated strong validation against orthogonal measurement methods, showing significant positive pan-cancer correlation with leukocyte fractions from DNA methylation profiles (r = 0.77) and strong negative correlations with tumor purities from both RNA-seq (ESTIMATE r = -0.83) and DNA-seq (ABSOLUTE r = -0.60) [1]. When compared against individual tools, iScores had the highest average correlations with original mixing fractions for all cell types deconvolved from pseudobulks [1].
Research Reagent Solutions for TME Deconvolution
Table 2: Essential Research Reagents and Resources for TME Deconvolution Studies
| Reagent/Resource | Function | Examples/Specifications | Application Notes |
|---|---|---|---|
| xCell 2.0 Algorithm | Cell type proportion estimation | Bioconductor package or web application; pre-trained references for human and mouse | Superior accuracy and spillover correction; can use custom references [4] |
| Reference scRNA-seq Datasets | Training and validation | Human Cell Atlas, Blueprint-Encode, tumor-specific references | Critical for accurate deconvolution; should match tissue type [4] [7] |
| Bulk RNA-seq Data | Primary input data | TCGA, GEO datasets, institutional cohorts | Requires proper normalization and quality control [1] |
| Orthogonal Validation Tools | Method verification | RNAScope/ImmunoFluorescence, immunohistochemistry, flow cytometry | Essential for benchmarking deconvolution accuracy [7] [6] |
| Spatial Transcriptomics | Spatial context validation | 10X Visium, Xenium platforms | Provides spatial distribution of cell types [7] |
| Computational Pathology Tools | Image-based validation | QuPath platform with object classifier | Enables single-cell annotation from H&E images [7] |
Workflow Diagrams for TME Deconvolution Analysis
xCell 2.0 Analytical Pipeline
xCell 2.0 Workflow
Integrated TME Analysis Framework
Integrated TME Analysis
Clinical Applications and Therapeutic Implications
Predictive Biomarker Discovery
TME deconvolution has demonstrated significant value in identifying predictive biomarkers for therapy response. In breast cancer, the DECODEM framework leveraging cellular deconvolution revealed that specific immune cells (myeloid, plasmablasts, B-cells) and stromal cells (endothelial, normal epithelial, cancer-associated fibroblasts) are highly predictive of chemotherapy response [8]. Ensemble models integrating the estimated expression of different cell types performed the best and outperformed models built on the original tumor bulk expression, highlighting the importance of comprehensive TME analysis [8].
Similarly, in acute myeloid leukemia (AML), a TIME-driven prognostic model constructed using xCell and ESTIMATE algorithms successfully stratified patients into high/low-risk groups with divergent survival (p-value = 0.00072) [5]. The model demonstrated predictive accuracy with AUC values of 63.38-68.5% for 1-5-year survival and revealed associations between high-risk scores and immunosuppressive cell subsets, including Tregs and M2 macrophages [5].
Drug Discovery and Target Identification
TME deconvolution enables novel approaches to drug discovery by identifying critical interactions within the tumor microenvironment. An immunoinformatic analysis of breast cancer TME identified five ligand-receptor pairs significantly associated with pathological stages and immune cell infiltration [9]. High expression of VEGFR2, TGFBR2 and TNFRSF12A in tumor tissue was positively correlated with increased overall survival, and these receptors varied significantly with nodal metastasis status and patient age groups [9]. This approach facilitated the identification of drug candidates that can disrupt these critical ligand-receptor interactions, providing novel insights for TME-directed therapy [9].
Validation Frameworks and Best Practices
Orthogonal Validation Strategies
Robust validation of deconvolution results requires multiple orthogonal approaches:
Computational Pathology: Machine learning-based computational tissue annotation (CTA) pipelines can provide high-resolution annotations on H&E-stained images, enabling validation of deconvolution results at single-cell resolution [7]. This approach has demonstrated strong agreement with molecular cell type markers from platforms like Xenium [7].
Multi-assay Benchmarking: Studies using multi-assay datasets from postmortem human prefrontal cortex have established frameworks for rigorous benchmarking of deconvolution algorithms against orthogonal measurements of cell type proportions with RNAScope/ImmunoFluorescence [6]. This approach identified Bisque and hspe as the most accurate methods for brain tissue analysis [6].
Spatial Transcriptomics Validation: Spatial transcriptomics technologies such as 10X Visium provide valuable validation platforms, though their spot-based resolution requires computational enhancement through paired H&E image analysis [7].
Method Selection Guidelines
Based on comprehensive benchmarking studies, method selection should consider:
Tissue Specificity: Performance varies significantly across tissues. Methods like Bisque and hspe perform best for brain tissue [6], while xCell 2.0 shows superior performance for immune cell deconvolution in cancer [4].
Cell Type Resolution: Most methods predict coarse-grained populations well, but show variable performance for fine-grained subpopulations [3]. xCell 2.0 shows improved performance for fine-grained immune cell states [4].
Integrated Approaches: For comprehensive TME analysis, integrating multiple deconvolution tools provides more robust results than any single method [1].
TME deconvolution, particularly through advanced implementations like xCell 2.0, has established itself as an essential tool in precision oncology. The ability to extract detailed cellular composition from bulk transcriptomics data enables researchers to leverage existing large-scale datasets while providing insights into TME heterogeneity that would be cost-prohibitive to obtain through single-cell methods alone. The clinical utility of these approaches has been demonstrated across multiple cancer types, with applications in prognosis, therapy response prediction, and biomarker discovery.
Future developments in this field will likely focus on improved integration of spatial information, enhanced resolution for fine-grained cell states, and standardized frameworks for clinical application. As validation methods continue to improve through computational pathology and multi-assay benchmarking, TME deconvolution will play an increasingly central role in translating complex microenvironmental interactions into actionable clinical insights.
The cellular heterogeneity of the tumor microenvironment (TME) plays a crucial role in cancer development, progression, and response to therapy. Understanding this complex cellular landscape is essential for advancing precision medicine in oncology. Bulk gene expression profiling has remained a common approach for studying the TME, particularly in clinical samples and large cohorts where single-cell RNA sequencing (scRNA-seq) may be prohibitively expensive or technically challenging. Computational deconvolution methods bridge this gap by inferring cellular composition from bulk transcriptomic data, enabling researchers to extract valuable insights about TME biology from existing and new datasets.
Signature-based deconvolution methods represent a powerful approach for characterizing cellular heterogeneity. These methods leverage cell-type-specific gene signatures to estimate relative abundances of different cell populations within complex tissue mixtures. Among these tools, xCell has gained significant popularity due to its high accuracy and ease of use. The recent introduction of xCell 2.0 marks a substantial evolution in signature-based deconvolution, addressing key limitations of its predecessor while introducing novel capabilities for TME analysis.
This article traces the technological evolution from xCell to xCell 2.0, detailing the methodological advances, benchmarking performance, and providing practical guidance for researchers seeking to apply these tools in cancer research and drug development.
The Original xCell Algorithm
Core Methodology and Applications
The original xCell algorithm was developed as a gene signature-based method for cell type enrichment analysis from bulk gene expression data. It employed a novel technique for reducing associations between closely related cell types, using spillover compensation to minimize false-positive signals from lineage-related populations. The method calculated single-sample Gene Set Enrichment Analysis (ssGSEA) scores for gene signatures and averaged scores across all signatures corresponding to specific cell types, providing enrichment scores for 64 immune and stromal cell types.
xCell gained widespread adoption in TME research due to its robust performance across diverse biological contexts. In application, xCell has demonstrated significant utility in characterizing the TME of various cancers. For instance, in triple-negative breast cancer (TNBC), researchers used xCell-derived scores of M2 macrophages, CD8+ T cells, and CD4+ memory T cells to construct a prognostic risk scoring system that effectively stratified patients into distinct survival groups [10]. The algorithm's ability to accurately portray cellular heterogeneity made it a valuable tool for exploring the relationship between TME composition and clinical outcomes.
Limitations and Need for Advancement
Despite its utility, the original xCell implementation had several constraints. It was pre-trained using specific reference gene expression datasets and could not be used with custom-made references, limiting its applicability to specific tissue types or experimental conditions. This was particularly problematic for TME studies, as tumors contain cell types not found in blood or normal tissues, making tissue-dedicated references essential for accurate deconvolution.
Additionally, the original xCell required manual identification of cell type dependencies to ensure that closely related cell types were not directly compared during signature generation. This labor-intensive process required substantial domain expertise and became increasingly challenging when dealing with references containing many cell types.
xCell 2.0: Technical Advancements and Methodological Improvements
Key Innovations and Algorithmic Enhancements
xCell 2.0 represents a significant evolution from the original algorithm, introducing several key innovations that enhance its flexibility, robustness, and performance [4] [11]. The most substantial advancement is the incorporation of a training function that enables users to utilize any reference dataset, including custom references tailored to specific research questions. This addresses a critical limitation of the original xCell and greatly expands the method's applicability across diverse biological contexts.
Table 1: Core Algorithmic Improvements in xCell 2.0
| Feature | xCell | xCell 2.0 |
|---|---|---|
| Reference Flexibility | Pre-trained references only | Custom references enabled via training function |
| Cell Type Dependency Handling | Manual identification required | Automated via ontological integration |
| Signature Generation | Fixed threshold criteria | Adaptive thresholds based on reference size |
| Spillover Correction | Manual control selection | Automatic identification of control cell types |
| Organism Support | Primarily human | Comprehensive human and mouse references |
The signature generation process in xCell 2.0 incorporates improved methodology for identifying differentially expressed genes, including automated handling of cell type dependencies and more robust signature generation [4]. A particularly important innovation is the introduction of ontological integration, where xCell 2.0 automatically extracts cell type lineage information directly from the standardized Cell Ontology (CL) [4]. This automation eliminates the need for manual intervention and ensures appropriate handling of lineage relationships during signature generation.
The threshold criteria for gene inclusion in signatures has been modified to accommodate references with variable numbers of cell types. While the original approach considered only genes that passed threshold criteria against the top three other cell types, xCell 2.0 implements a threshold-based approach requiring genes to pass criteria against at least 50% of cell types in the reference [4]. This adaptation ensures robust signature generation across diverse reference datasets.
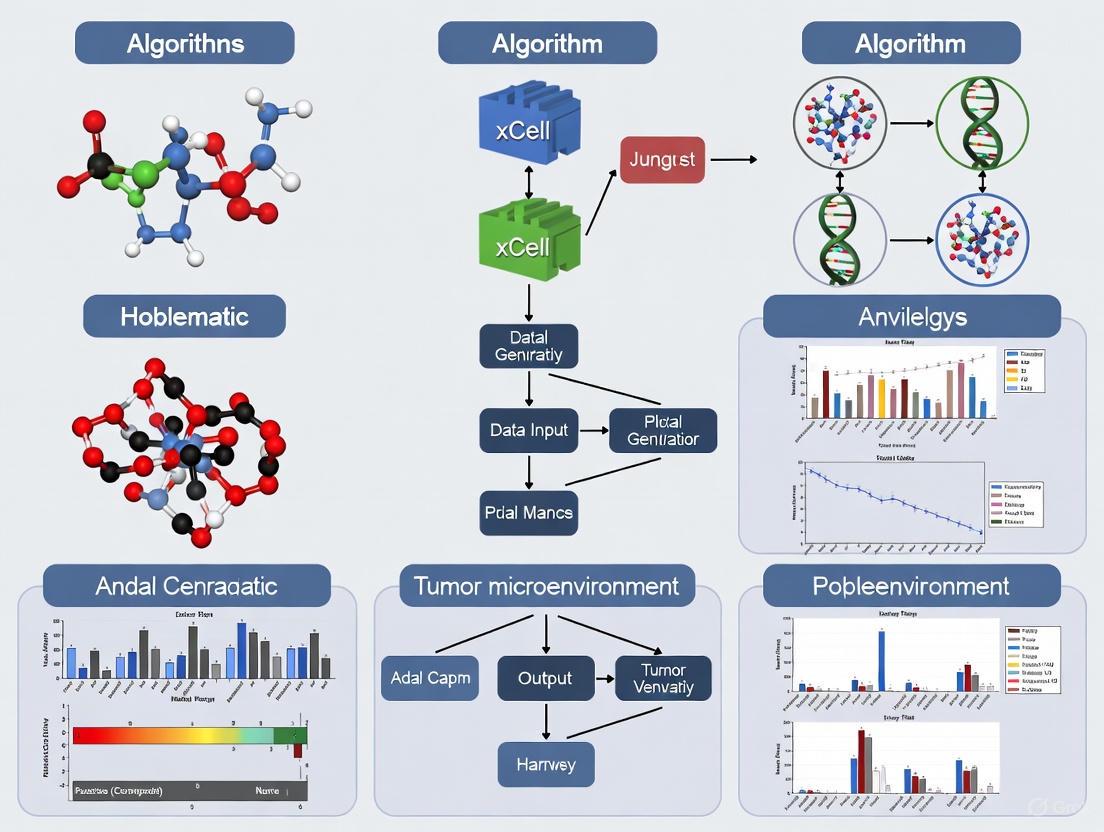
Workflow and Implementation
The xCell 2.0 pipeline employs a structured workflow for generating custom reference objects and performing cell type enrichment analysis [12]. The process begins with obtaining a reference gene expression dataset of pure cell types, which can be derived from microarray, bulk RNA-seq, or scRNA-seq data. The algorithm then generates cell type gene signatures using an improved approach that compares gene expression quantiles between cell types to identify differentially expressed genes.
Diagram 1: xCell 2.0 Training Workflow. The process for creating custom reference objects involves four key steps, from data preparation to parameter learning.
For practical implementation, xCell 2.0 is available as an R package through Bioconductor and GitHub, providing both programmatic access and a locally hosted web application [12]. The package includes comprehensive documentation and vignettes to facilitate adoption by researchers with varying levels of computational expertise.
Performance Benchmarking and Validation
Comparative Evaluation Against Other Methods
The performance of xCell 2.0 has been rigorously evaluated through extensive benchmarking against other deconvolution methods. In a comprehensive assessment using nine human and mouse reference sets and 26 validation datasets encompassing 1,711 samples and 67 cell types, xCell 2.0 outperformed all eleven other tested methods across distinct reference datasets [4] [11]. The algorithm demonstrated superior accuracy and consistency across diverse biological contexts, with particular strength in minimizing spillover effects between related cell types.
xCell 2.0 was further validated using the independent Deconvolution DREAM Challenge dataset, a community-wide benchmark that evaluated both published and newly developed deconvolution methods [3]. The Challenge focused on predicting both coarse-grained populations (eight major immune and stromal cell types) and fine-grained subpopulations (14 specific cell states), using in vitro and in silico transcriptional profiles of admixed cancer and healthy immune cells as ground truth [3].
Table 2: Performance Comparison of Deconvolution Methods
| Method | Overall Accuracy | Spillover Control | Fine-Grained Resolution | TME Application |
|---|---|---|---|---|
| xCell 2.0 | Superior | Best performance | High | Excellent |
| BayesPrism | High | Good | High | Excellent |
| Scaden | High | Moderate | Medium | Good |
| MuSiC | High | Moderate | Medium | Good |
| DWLS | Medium | Good | Medium | Good |
| CIBERSORTx | Medium | Moderate | Medium | Good |
| EPIC | Low | Poor | Low | Limited |
In the context of TME deconvolution, a separate benchmarking study focused specifically on breast cancer using scRNA-seq simulated bulk mixtures revealed important considerations for method selection [13]. This study evaluated nine TME deconvolution methods, including BayesPrism, Scaden, CIBERSORTx, MuSiC, DWLS, and others, assessing their performance across variable tumor purity levels. The findings indicated that methods perform differently depending on tumor purity, with some showing improved performance in high-purity samples while others performed better in low-purity contexts [13].
Clinical and Translational Applications
The clinical utility of xCell 2.0 was demonstrated in a pan-cancer immune checkpoint blockade (ICB) response prediction study [4]. When applied to bulk RNA-seq data from 2,007 cancer patients prior to ICB treatment across different cancer types, xCell 2.0-derived TME features significantly improved prediction accuracy compared to models using only cancer type and treatment information. The method outperformed other deconvolution approaches and established prediction scores, highlighting its potential for advancing precision immuno-oncology.
In another translational application, researchers successfully employed xCell (the original version) as part of a multiomics integration analysis to identify tumor cell-derived macrophage migration inhibitory factor (MIF) as a therapeutic target in osteosarcoma [14]. The xCell algorithm was used to evaluate immune cell infiltration and activity, contributing to the identification of MIF as a key regulator of macrophage polarization and chemotaxis. This finding was subsequently validated through functional assays, demonstrating the practical utility of cell type enrichment analysis in target discovery.
Practical Implementation Guide
Research Reagent Solutions
xCell 2.0 provides researchers with a comprehensive toolkit for TME deconvolution, including both pre-trained references and the capability to generate custom references specific to research needs.
Table 3: Essential Research Reagents and Resources for xCell 2.0
| Resource Type | Examples | Function | Availability |
|---|---|---|---|
| Pre-trained References | BlueprintEncode, ImmGenData, LM22, Pan Cancer | Ready-to-use references for common research contexts | https://dviraran.github.io/xCell2refs |
| Single-cell References | Tabula Muris Blood, Tabula Sapiens Blood, Pan Cancer | High-resolution references from scRNA-seq data | Public repositories + xCell2 collection |
| Software Package | xCell2 R package | Core algorithm implementation | Bioconductor/GitHub |
| Web Application | Local hosted web tool | User-friendly interface for analysis | Included with package |
Experimental Protocol for xCell 2.0 Analysis
Protocol 1: Creating a Custom Reference with xCell2Train
- Input Preparation: Prepare a reference gene expression matrix with genes in rows and samples/cells in columns, normalized for both gene length and library size. The data can be in linear or logarithmic space.
- Labels Data Frame Creation: Create a data frame with four required columns: "ont" (Cell Ontology ID or NA), "label" (cell type name), "sample" (identifier matching reference matrix), and "dataset" (source identifier).
- Reference Generation: Execute the training function:
- Validation: Assess reference quality using built-in diagnostics and save the object for future use.
Protocol 2: Cell Type Enrichment Analysis with xCell2Analysis
- Data Preparation: Format bulk gene expression data with genes in rows and samples in columns, using the same gene identifiers as the reference.
- Reference Selection: Choose an appropriate pre-trained or custom reference object matching the biological context.
- Execute Analysis:
- Result Interpretation: The output is a matrix of enrichment scores with cell types as rows and samples as columns. Higher scores indicate stronger presence of cell types. Scores are relative and most useful for comparative analysis across samples.
Diagram 2: xCell 2.0 Analysis Workflow. The process for performing cell type enrichment analysis from bulk expression data, culminating in various downstream applications.
Best Practices and Troubleshooting
For optimal results with xCell 2.0, researchers should:
- Select references that closely match the tissue type and biological context of their study
- Ensure sufficient overlap between genes in the bulk data and reference
- Use spillover correction for closely related cell types but verify it doesn't introduce new biases
- Interpret results as relative enrichment scores rather than absolute proportions
- Validate critical findings with orthogonal methods when possible
Common issues and solutions include:
- Low gene overlap: Check gene identifiers and consider mapping to standard nomenclatures
- Poor performance: Try alternative references or validate with known positive controls
- Unexpected scores: Examine raw expression of marker genes to contextualize results
The evolution from xCell to xCell 2.0 represents significant progress in signature-based deconvolution methods for TME analysis. By addressing key limitations of the original algorithm—particularly through enabling custom reference generation and automating cell type dependency handling—xCell 2.0 has expanded the applicability and robustness of cell type enrichment analysis across diverse research contexts.
The demonstrated performance of xCell 2.0 in comprehensive benchmarking studies, combined with its successful application in predicting response to immune checkpoint blockade, underscores its value as a tool for both basic research and translational applications. As single-cell technologies continue to generate increasingly detailed references of cellular heterogeneity in health and disease, the flexible framework of xCell 2.0 positions it to leverage these resources for continued improvement in deconvolution accuracy.
For the research community, xCell 2.0 offers a versatile and powerful platform for interrogating cellular heterogeneity from bulk transcriptomic data. Its integration with Bioconductor, comprehensive documentation, and collection of pre-trained references lower barriers to adoption, while its training functionality enables customization for specialized applications. As precision medicine continues to emphasize the importance of TME composition in therapeutic response, tools like xCell 2.0 will play an increasingly vital role in extracting maximal biological insight from transcriptomic data.
The digital dissection of the tumor microenvironment (TME) represents a cornerstone of modern cancer research, enabling the quantification of cellular heterogeneity from bulk transcriptomic data. Three interconnected computational techniques form the critical foundation for these analyses: Single-Sample Gene Set Enrichment Analysis (ssGSEA), Spillover Compensation, and Linear Transformation. When integrated within algorithms such as xCell, these methods empower researchers to transform complex bulk RNA-sequencing data into actionable insights about the relative abundance of immune and stromal cell populations within the TME [15] [4] [16]. This framework is particularly vital for translational applications, including prognostic model development and predicting response to immune checkpoint blockade therapy [17] [4] [10]. The following application notes detail the core mechanics, experimental protocols, and practical implementation of these methodologies to ensure robust, reproducible, and biologically meaningful TME analysis.
Core Algorithmic Components and Their Functions
Single-Sample Gene Set Enrichment Analysis (ssGSEA)
ssGSEA is an extension of Gene Set Enrichment Analysis (GSEA) that calculates a separate enrichment score for each sample and gene set pair, effectively quantifying the activity of a biological process or the abundance of a cell type within an individual sample [18] [19]. Unlike standard GSEA, which requires multiple samples per group for comparison, ssGSEA operates on a single sample, making it ideal for clinical datasets where sample numbers may be limited [17]. The algorithm works by ranking all genes in a single sample by their expression level, then evaluating the distribution of a predefined gene set within this ranked list using a Kolmogorov-Smirnov-like random walk statistic [19]. The resulting enrichment score (ES) represents the degree to which the genes in the signature are collectively overexpressed at one end of the ranked list. This score is then normalized to generate a normalized enrichment score (NES), which allows for comparison across different gene sets and samples [19]. In the context of TME deconvolution, these gene signatures are curated to represent specific immune or stromal cell types, and their enrichment scores serve as proxies for cell abundance [17] [18].
Spillover Compensation
Spillover Compensation addresses a critical challenge in cellular deconvolution: the high transcriptional similarity between closely related cell types (e.g., CD4+ T cells and CD8+ T cells) [15] [4]. This similarity can cause a "spillover" effect, where the gene signature for one cell type also captures signals from a related cell type, leading to inaccurate abundance estimates [16]. The xCell algorithm implements a dedicated spillover compensation technique that leverages in-silico simulations of cell type mixtures to model and correct for these dependencies [15]. The process generates a spillover matrix that quantifies the pairwise interference between all cell types. A spillover correction strength parameter (α) is then applied, allowing users to balance the correction of genuine spillover effects against the risk of over-correction [4] [16]. In xCell 2.0, this process has been enhanced through the automated identification of lineage relationships between cell types using the Cell Ontology (CL), eliminating the need for manual, expert-led identification of these dependencies [4].
Linear Transformation
Linear Transformation is a mathematical operation applied to convert the non-linear enrichment scores generated by ssGSEA into a linear scale that better approximates actual cell type proportions [15] [16]. The raw ssGSEA enrichment scores are not linearly related to cell abundance, which limits their direct interpretability and comparability across different cell types [15]. By applying a linear transformation—learned from in-silico mixtures of pure cell types—xCell translates these enrichment scores into scores that show a linear relationship with the known fractions of cell types in the simulated mixtures [15]. This transformation is fundamental to producing final scores that allow for meaningful comparison of abundances not just across samples, but also across different cell types within the same sample [15] [20].
The diagram below illustrates the integrated workflow of these three components within the xCell algorithm.
Quantitative Performance and Validation Data
The integration of ssGSEA, spillover compensation, and linear transformation within xCell 2.0 has been rigorously validated against other deconvolution methods. The following tables summarize key quantitative findings from these benchmark studies.
Table 1: Impact of Spillover Correction Strength (α) on Estimation Accuracy in xCell 2.0 [4] [16]
| Correction Strength (α) | Direct Correlation (Mean Pearson r) | Spill Correlation (Mean Pearson r) |
|---|---|---|
| 0.0 (No correction) | 0.72 | 0.58 |
| 0.2 | 0.71 | 0.45 |
| 0.4 | 0.71 | 0.35 |
| 0.6 | 0.70 | 0.28 |
| 0.8 | 0.70 | 0.22 |
| 1.0 (Full correction) | 0.69 | 0.18 |
Table 2: Benchmarking Performance of xCell 2.0 Against Other Methods Across 26 Validation Datasets [4] [16]
| Deconvolution Method | Average Overall Accuracy (Pearson r) | Performance in Minimizing Spillover | Consistency Across Platforms |
|---|---|---|---|
| xCell 2.0 | 0.75 | Best | Best |
| xCell (original) | 0.71 | Good | Good |
| CIBERSORT | 0.68 | Moderate | Moderate |
| Other methods (n=9) | <0.65 | Variable | Variable |
Table 3: Prognostic Value of Immune-Related Gene Signatures Derived via ssGSEA in OSCC [17]
| Risk Group | 5-Gene Signature Model | Overall Survival (Hazard Ratio) | Immune Checkpoint Gene Expression |
|---|---|---|---|
| Low-Risk | CCL18, CXCL13, HLA-DOB, HLA-DPB2, TNFRSF17 | Reference (1.0) | Lower |
| High-Risk | CCL18, CXCL13, HLA-DOB, HLA-DPB2, TNFRSF17 | 2.45 (p < 0.001) | Higher |
Experimental Protocols
Protocol 1: Generating a Custom Reference Object for xCell 2.0
Purpose: To create a custom reference object for cell type enrichment analysis using xCell 2.0, enabling tailored investigation of specific tissues or disease contexts [4] [16].
Workflow Overview:
Materials:
- Hardware: Computer with at least 8GB RAM and multi-core processor
- Software: R environment (v4.0 or higher), xCell 2.0 package installed from Bioconductor
- Input Data: A reference gene expression dataset of pure cell types (microarray, bulk RNA-seq, or scRNA-seq data)
Procedure:
- Reference Data Input: Provide a gene expression matrix of pure cell types as input. The matrix should have genes as rows and samples as columns, with sample annotations clearly specifying the cell type for each column [4].
- Automated Gene Signature Generation: Run the signature generation function. xCell 2.0 will automatically:
- Handle Cell Type Dependencies: Use Cell Ontology (CL) IDs to identify lineage relationships and avoid comparing closely related cell types during signature generation [4] [16].
- Apply Threshold Criteria: Identify differentially expressed genes for each cell type against at least 50% of other cell types in the reference, creating hundreds of signatures per cell type using various expression percentile thresholds [16].
- Parameter Learning via In-Silico Simulation: The algorithm will automatically:
- Generate synthetic expression profiles by mixing the cell type of interest with a control cell type (automatically selected as the most distinct cell type based on gene expression correlation) [16].
- Fit a linear formula to transform raw ssGSEA enrichment scores to values that are linearly correlated with cell type abundance in the simulated mixtures [15] [16].
- Spillover Matrix Calculation: The algorithm calculates pairwise spillover effects between all cell types (excluding those with lineage dependencies) to create a spillover matrix [4].
- Output: The process yields a custom reference object containing the gene signatures, linear transformation parameters, and spillover matrix, ready for deconvolution of bulk datasets [4].
Protocol 2: TME Deconvolution and Prognostic Model Construction
Purpose: To deconvolute the cellular composition of tumor samples and construct a prognostic model based on key immune cell populations, as applied in triple-negative breast cancer (TNBC) and other malignancies [10] [17].
Materials:
- Input Data: Bulk tumor gene expression data (e.g., from TCGA or GEO) from the cancer of interest
- Software: R packages:
xCell2,survival,randomForestSRC,timeROC[10] - Reference: Pre-trained xCell 2.0 reference object (e.g., the pan-cancer immune reference)
Procedure:
- Run xCell 2.0 Deconvolution:
- Input the bulk tumor gene expression matrix into the xCell 2.0 algorithm along with the chosen reference object.
- Execute the analysis to obtain enrichment scores for 64 immune and stromal cell types for each tumor sample [10].
- Identify Prognosis-Related Cells:
- Perform univariate Cox regression analysis on each cell type's xCell score against overall survival data.
- Select cells with a statistically significant association with survival (p-value < 0.05) for further modeling [10].
- Construct a Random Survival Forest (RSF) Model:
- Input the significant cell types into an RSF model.
- Use the variable importance measure from the RSF to identify the most potent prognostic cell subsets [10].
- Define Risk Groups and Validate:
- Use the RSF model to stratify patients into distinct risk groups based on key cell type combinations (e.g., M2 macrophages, CD8+ T cells, CD4+ memory T cells) [10].
- Validate the prognostic performance of the risk groups in one or more independent validation cohorts using Kaplan-Meier survival analysis and log-rank tests [17] [10].
- Assess the model's predictive accuracy using time-dependent Receiver Operating Characteristic (ROC) analysis [10].
Table 4: Key Research Reagents and Computational Tools for TME Deconvolution
| Resource Name | Type | Function/Purpose | Availability |
|---|---|---|---|
| xCell 2.0 | Software Package | Performs cell type enrichment analysis from bulk gene expression data using ssGSEA, linear transformation, and spillover compensation. | Bioconductor |
| Pre-trained Reference Objects | Data Resource | Curated collections of gene signatures for human and mouse cell types, enabling immediate analysis without custom training. | https://dviraran.github.io/xCell2refs [4] [16] |
| TCGA (The Cancer Genome Atlas) | Data Resource | Provides bulk RNA-seq data and clinical information for thousands of tumor samples, serving as a primary source for discovery and validation. | https://portal.gdc.cancer.gov [17] [19] |
| Cell Ontology (CL) | Ontology | A structured, controlled vocabulary for cell types, used by xCell 2.0 to automatically identify lineage relationships and manage cell type dependencies. | http://www.obofoundry.org/ontology/cl.html [4] |
| ssGSEA 2.0 Script | Algorithm | The core script for calculating single-sample GSEA scores, available from the Broad Institute. | https://github.com/broadinstitute/ssGSEA2.0 [19] |
The accurate deconvolution of bulk gene expression data to determine cellular heterogeneity is fundamental to advancing our understanding of the tumor microenvironment (TME). xCell 2.0 represents a significant evolution in computational tools for cell type proportion estimation, introducing critical features that address specific challenges in TME analysis. This upgraded version builds upon the original xCell methodology, which gained widespread adoption due to its high accuracy and ease of use, but was limited by its pre-trained nature and inability to accommodate custom references tailored to specific tissue types or experimental conditions [4] [15].
For researchers focusing on the complex cellular landscape of the TME, the inability to use tissue-dedicated references presented a substantial limitation, as the TME contains cell types not found in standard blood-based references [4] [16]. xCell 2.0 directly addresses this constraint through a redesigned architecture that incorporates a training function, enabling researchers to utilize any reference dataset—including single-cell RNA-seq data—specific to their research context [12]. This flexibility, combined with improved signature generation and automated handling of cell type dependencies, positions xCell 2.0 as a versatile and robust tool for TME investigation across diverse cancer types and research applications.
Core Technical Advancements in xCell 2.0
Automated Ontological Integration for Cell Type Dependencies
A fundamental challenge in cellular deconvolution is properly handling lineage relationships between cell types, where closely related cell types (e.g., T cells and CD4+ T cells) can exhibit similar gene expression patterns, leading to "spillover" effects that compromise accuracy. The original xCell algorithm required manual identification of these dependencies—a labor-intensive process requiring substantial domain expertise that became increasingly impractical with custom references containing numerous cell types [4] [16].
xCell 2.0 introduces automated ontological integration to resolve this limitation. The algorithm now automatically extracts cell type lineage information directly from the standardized Cell Ontology (CL), enabling the pipeline to account for cell type dependencies without manual intervention [4] [16]. This implementation ensures that closely related cell types are not directly compared during signature generation, significantly improving the specificity of cell type estimates. Benchmark validation studies demonstrate that this automated handling of dependencies substantially enhances overall signature performance compared to methods that ignore these critical biological relationships [4].
Custom Reference Training Capability
The most transformative advancement in xCell 2.0 is its capacity for generating custom reference objects, which dramatically expands its applicability across diverse research contexts. The xCell2Train function enables researchers to create tailored reference objects using their own transcriptomic data from various platforms, including microarray, bulk RNA-seq, or single-cell RNA-seq [12]. This functionality addresses a critical need in TME research, where tissue-specific and context-specific references are essential for accurate cellular deconvolution.
The custom reference training process incorporates several technical improvements:
- Adaptive threshold criteria: Instead of the original approach of comparing against the top three other cell types, xCell 2.0 implements a threshold-based approach requiring genes to pass differential expression criteria against at least 50% of cell types in the reference [4]. This modification accommodates the variability in cell type numbers across custom references while maintaining stringency.
- Robust signature generation: Hundreds of signatures for each cell type are generated using various predefined thresholds, including percentiles of gene expression, expression differences between cell types, and boundaries on gene numbers per signature [4].
- Automated parameter learning: xCell 2.0 automatically generates in-silico simulations to learn parameters that transform enrichment scores to linear proportions and correct for spillover effects, selecting the most distinct cell type as control automatically [4].
Enhanced Performance in Minimizing Spillover Effects
Spillover effects—where signatures of closely related cell types show correlation—have been a persistent challenge in deconvolution algorithms. xCell 2.0 introduces a refined spillover correction system that allows researchers to control correction strength through the α parameter [4]. This controlled correction enables balancing between genuine spillover correction and potential over-correction that could introduce new biases. Validation experiments demonstrate that while direct correlation between estimated and true proportions remains stable across α values, spill correlation (correlation with similar cell types) decreases significantly with stronger correction, indicating enhanced specificity [4].
Table 1: Key Technical Improvements in xCell 2.0 Compared to Original xCell
| Feature | Original xCell | xCell 2.0 | Impact on TME Research |
|---|---|---|---|
| Reference flexibility | Pre-trained references only | Custom references from any dataset | Enables tissue-specific TME analysis |
| Dependency handling | Manual identification | Automated via Cell Ontology | Reduces bias in complex cellular mixtures |
| Signature generation | Fixed thresholds | Adaptive thresholds (50% of cell types) | Improved performance across diverse references |
| Spillover correction | Fixed parameters | Adjustable strength (α parameter) | Enhanced specificity for related cell types |
| Platform compatibility | Limited platforms | Microarray, RNA-seq, scRNA-seq | Broad applicability across experimental designs |
Experimental Protocols for xCell 2.0 Implementation
Protocol 1: Generating Custom Reference Objects
The creation of custom reference objects represents a foundational workflow in xCell 2.0 application for TME studies. The following step-by-step protocol details this process:
Step 1: Input Data Preparation Prepare two essential inputs:
- Reference gene expression matrix: Can be derived from microarray, bulk RNA-Seq, or single-cell RNA-Seq data with genes in rows and samples/cells in columns. Data should be normalized for both gene length and library size, and can be in either linear or logarithmic space [12].
- Labels data frame: Must contain four columns with specific information about each sample/cell:
- "ont": Cell type ontology identifier (e.g., "CL:0000545" or NA if not applicable)
- "label": Cell type name (e.g., "T-helper 1 cell")
- "sample": Sample/cell identifier matching column names in the reference matrix
- "dataset": Source dataset or subject identifier [12]
Step 2: Reference Object Generation Execute the training function with properly formatted inputs:
The algorithm automatically processes the data through ontological integration, signature generation, and parameter learning for spillover correction [12].
Step 3: Validation and Storage Validate the resulting reference object and store for future use. The complete process typically requires several hours depending on reference size and computational resources.
Diagram 1: Custom Reference Creation Workflow. This diagram illustrates the automated process for generating custom xCell2 reference objects, highlighting key steps from data preparation to final reference object.
Once a custom reference object is generated or selected, researchers can perform cell type enrichment analysis on bulk transcriptomics data using the following protocol:
Step 1: Data Preparation
- Prepare bulk gene expression matrix from tumor samples with genes in rows and samples in columns
- Ensure proper normalization and formatting compatible with the reference object
- Load the appropriate xCell2 reference object (custom or pre-trained)
Step 2: Execute Enrichment Analysis Run the analysis function with required parameters:
Step 3: Results Interpretation The function returns a matrix of cell type enrichment scores where:
- Rows represent cell types
- Columns represent samples from the input mixture
- Higher scores indicate stronger presence of that cell type
- Scores are relative rather than absolute proportions, enabling comparison across samples but not direct quantification of absolute cell numbers [12]
Step 4: Downstream Analysis
- Correlate cell type enrichment scores with clinical variables
- Perform differential enrichment analysis between sample groups
- Integrate scores as features in machine learning models for outcome prediction
Performance Benchmarking and Validation
Comprehensive Benchmarking Against Established Methods
xCell 2.0 has undergone rigorous validation against current state-of-the-art deconvolution methods. In comprehensive benchmarking involving eleven popular deconvolution tools across nine human and mouse reference sets and 26 validation datasets (encompassing 1711 samples and 67 cell types), xCell 2.0 demonstrated superior accuracy and consistency across diverse biological contexts [4] [16]. The algorithm also showed the best performance in minimizing spillover effects between related cell types, a critical advantage for resolving closely related immune subsets in the TME.
Additional validation using the independent Deconvolution DREAM Challenge dataset confirmed xCell 2.0's robust performance [4]. This extensive evaluation establishes xCell 2.0 as a leading tool for cellular deconvolution, particularly valuable for the complex cellular mixtures characteristic of tumor microenvironments.
Table 2: Research Reagent Solutions for xCell 2.0 Implementation
| Resource Type | Specific Examples | Application Context | Access Method |
|---|---|---|---|
| Pre-trained human references | BlueprintEncode, Immune Compendium, LM22, Pan Cancer, Tabula Sapiens Blood | General human TME studies | Built-in package data or download from project website |
| Pre-trained mouse references | ImmGenData, MouseRNAseqData, Tabula Muris Blood | Murine model systems | Built-in package data or download from project website |
| Custom reference training data | DICE database, scRNA-seq datasets | Tissue-specific or novel cell type analysis | xCell2Train() function with user data |
| Analysis workflows | xCell2Analysis() function | Standard enrichment analysis | Direct implementation in R |
| Validation datasets | Deconvolution DREAM Challenge, synthetic mixtures | Method verification and benchmarking | Public repository sources |
Application in Predictive Oncology
The translational potential of xCell 2.0 is particularly evident in its application to immunotherapy response prediction. In a pan-cancer evaluation involving bulk RNA-seq data from 2007 cancer patients prior to treatment with immune checkpoint blockade (ICB), xCell 2.0-derived TME features significantly improved prediction accuracy compared to models using only cancer type and treatment information [4] [16]. Furthermore, xCell 2.0 outperformed other deconvolution methods and established prediction scores, highlighting its potential for advancing precision immuno-oncology.
In a separate study focused on acute myeloid leukemia (AML), xCell 2.0 was instrumental in constructing a tumor immune microenvironment-driven prognostic model that successfully stratified patients into high and low-risk groups with divergent survival outcomes (p-value = 0.00072) [5]. The model demonstrated predictive accuracy with AUC values of 63.38–68.5% for 1–5-year survival, and revealed clinically relevant associations between high-risk scores and immunosuppressive cell subsets, including Tregs and M2 macrophages [5].
Implementation Guidelines and Best Practices
Reference Selection Strategy
Choosing appropriate references is critical for successful TME deconvolution. xCell 2.0 provides multiple pre-trained references covering various tissues and organisms, but also supports custom reference generation. The following decision workflow guides appropriate reference selection:
Diagram 2: Reference Selection Decision Framework. This diagram provides a strategic approach for researchers to select the most appropriate reference type for their specific TME study, balancing between pre-trained options and custom reference creation.
Practical Implementation Considerations
Data Quality Requirements Successful application of xCell 2.0 depends on several data quality factors:
- Reference data should represent pure cell populations with accurate annotation
- Bulk expression data should undergo standard normalization procedures
- Sufficient sequencing depth is required for detection of rare cell populations
- Batch effects should be minimized when integrating multiple datasets
Computational Resources
- Custom reference generation is computationally intensive and benefits from multi-threading
- Analysis of large bulk datasets typically requires moderate memory allocation
- The R package implementation facilitates integration with bioinformatics workflows
Interpretation Guidelines
- Results represent relative enrichment rather than absolute cell counts
- Cross-sample comparisons are valid, but cross-cell type comparisons require caution
- Biological context should inform interpretation of cell type enrichment scores
- Integration with orthogonal validation methods (e.g., immunohistochemistry) strengthens conclusions
xCell 2.0 represents a substantial advancement in computational tools for TME deconvolution, addressing critical limitations of previous methods through automated ontological integration and custom reference training capabilities. These features enable researchers to tailor analyses to specific tissue contexts and cancer types, providing unprecedented flexibility for tumor microenvironment research. The robust performance of xCell 2.0 in benchmark evaluations and its demonstrated utility in predicting response to immunotherapy underscore its value as a tool for both basic cancer biology and translational research.
The implementation of xCell 2.0 in standardized protocols, as outlined in this article, provides researchers with a clear pathway to leverage these advancements in their own TME studies. As single-cell technologies continue to generate increasingly comprehensive reference datasets, the capacity to incorporate these resources into deconvolution frameworks through tools like xCell 2.0 will be essential for maximizing their utility in both retrospective analyses of existing bulk data and prospective study designs. The continued development and refinement of computational deconvolution methods represents a critical frontier in cancer research, enabling increasingly precise characterization of the cellular ecosystems that govern tumor behavior and therapeutic response.
Cellular heterogeneity within the tumor microenvironment (TME) is a critical determinant of cancer progression, therapeutic response, and patient outcomes. The xCell algorithm represents a transformative bioinformatics approach for digitally dissecting this complexity by estimating the enrichment of diverse cell types from bulk gene expression data. Unlike conventional methods that focus on limited immune populations, xCell provides an unprecedented resolution of 64 immune and stromal cell types, offering researchers a comprehensive tool for TME analysis. This capability is particularly valuable in oncology research, where understanding the cellular composition of tumors can reveal predictive biomarkers and inform therapeutic strategies [21] [10].
The fundamental innovation of xCell lies in its gene signature-based methodology, which was learned from thousands of pure cell types from various sources. By applying a novel technique for reducing associations between closely related cell types, xCell allows researchers to reliably portray the cellular heterogeneity landscape of tissue expression profiles. This approach has demonstrated superior performance compared to previous methods when validated through both in-silico simulations and cytometry immunophenotyping [22]. The recent introduction of xCell 2.0 has further enhanced these capabilities with a training function that permits utilization of any reference dataset, automated handling of cell type dependencies, and more robust signature generation [4] [11].
For researchers and drug development professionals, xCell offers a powerful means to leverage existing bulk transcriptomic data from sources like The Cancer Genome Atlas (TCGA) to gain insights into cellular dynamics that would otherwise require expensive single-cell technologies. This is particularly relevant for retrospective studies and clinical trial analyses where fresh tissue for single-cell RNA sequencing is unavailable [4]. The comprehensive cell type coverage encompassing 34 immune cells, 13 stromal cells, 9 stem cells, and 8 other cells provides an unmatched detailed view of the TME components that influence cancer behavior and treatment response [21].
Algorithm Evolution: From xCell to xCell 2.0
Technical Advancements and Improved Performance
xCell 2.0 represents a significant evolution from its predecessor, introducing architectural improvements that enhance its accuracy, flexibility, and applicability across diverse research contexts. The key advancement in xCell 2.0 is its genericity—users can now utilize any reference, including single-cell RNA-Seq data, to train a custom xCell2 reference object for analysis [12]. This addresses a critical limitation of the original xCell, which was pre-trained using reference gene expression datasets and could not be used with custom-made references, limiting its usability for specific tissue types or experimental conditions [4].
The updated algorithm incorporates several technical innovations that contribute to its enhanced performance. xCell 2.0 introduces ontological integration that automates the identification of lineage relationships among cell types using standardized Cell Ontology (CL) identifiers. This automation eliminates the labor-intensive manual identification of cell type dependencies required in the original version, ensuring that closely related cell types (e.g., T cells and CD4+ T cells) are not directly compared during signature generation, thereby reducing lineage-related biases [4]. Additionally, xCell 2.0 modifies the threshold criteria for determining gene inclusion into signatures, implementing a threshold-based approach of at least 50% of cell types in the reference rather than just the top three other cell types. This change accommodates variability in the number of cell types in custom references while maintaining robust signature generation [4].
Comprehensive benchmarking demonstrates xCell 2.0's superior performance relative to other deconvolution methods. When evaluated against eleven popular deconvolution methods using nine human and mouse reference sets and 26 validation datasets encompassing 1711 samples and 67 cell types, xCell 2.0 outperformed all other tested methods across distinct reference datasets. It also showed the best performance in minimizing spillover effects between related cell types—a common challenge in deconvolution algorithms [4] [11]. The algorithm's robustness was further validated using the independent Deconvolution DREAM Challenge dataset, confirming its consistent accuracy across diverse biological contexts [4].
Workflow and Computational Architecture
The xCell 2.0 pipeline employs a sophisticated multi-step process for generating custom reference objects used in cell type enrichment analysis. The workflow begins with obtaining a reference gene expression dataset of pure cell types, which can be derived from microarray, bulk RNA-seq, or scRNA-Seq data. The algorithm then generates cell type gene signatures by comparing gene expression quantiles between cell types to identify differentially expressed genes, while automatically accounting for lineage relationships through ontological integration [4].
In the signature generation phase, xCell 2.0 creates hundreds of signatures for each cell type using various predefined thresholds, including different percentiles of gene expression, the difference in expression between the cell type of interest and others, and the minimum and maximum number of genes per signature. Finally, the algorithm generates in-silico simulations to learn parameters that transform enrichment scores to linear scores and correct for spillover. These simulations are performed with automatic identification of control cell types, eliminating the need for manual intervention [4].
Table 1: Key Improvements in xCell 2.0
| Feature | Original xCell | xCell 2.0 |
|---|---|---|
| Reference Flexibility | Pre-trained references only | Custom references from any dataset |
| Cell Type Dependency Handling | Manual identification | Automated ontological integration |
| Signature Generation | Comparison against top 3 cell types | Threshold of 50% of cell types |
| Spillover Correction | Manual control selection | Automatic control identification |
| Validation Performance | High accuracy | Superior to 11 other methods |
The computational implementation of xCell 2.0 is available as a Bioconductor-compatible R package, equipped with a large collection of pre-trained cell type signatures for human and mouse research. The package includes comprehensive functionality for both training custom references and performing cell type enrichment analysis on bulk transcriptomics data [12]. For accessibility, it is also provided via a locally hosted web application, ensuring researchers with varying computational expertise can leverage its capabilities [4] [11].
Figure 1: xCell 2.0 Analytical Workflow. The diagram illustrates the key steps in creating custom reference objects and performing cell type enrichment analysis.
Research Applications in Tumor Microenvironment Analysis
Breast Cancer Cellular Heterogeneity Mapping
In a comprehensive study of breast cancer TME, researchers applied xCell to create a cellular heterogeneity map of 1,092 breast tumor and adjacent normal tissues from TCGA. The analysis revealed significant differences in cell fractions between tumor and normal tissues, with tumors displaying higher proportions of immune cells, including CD4+ Tem, CD8+ naïve T cells, and CD8+ Tcm [21]. This large-scale application demonstrated xCell's capability to handle substantial sample sizes while maintaining sensitivity to detect nuanced cellular differences.
The breast cancer study further identified 28 cell types significantly associated with overall survival in univariate analysis. Specifically, CD4+ Tem, CD8+ Tcm, CD8+ T-cells, CD8+ naive T-cells, and B cells emerged as positive prognostic factors, while CD4+ naive T-cells represented negative prognostic factors for breast cancer patients [21]. The research also uncovered coordinated expression of immune inhibitory receptors (PD1, CTLA4, LAG3, and TIM3) on specific T-cell subsets in breast tumors, with PD1 and CTLA4 both positively correlated with CD8+ Tcm and CD8+ T cells. These findings illustrate how xCell-derived cell enrichment scores can reveal clinically relevant immune patterns within the TME [21].
Striking differences in cellular heterogeneity were discovered among different breast cancer subtypes defined by Her2, ER, and PR status. Triple-negative patients exhibited the highest fraction of immune cells while luminal type patients showed the lowest, suggesting distinct immune microenvironments across molecular subtypes that may influence therapy response [21]. This application highlights xCell's utility in stratifying patients based on TME characteristics, potentially guiding personalized treatment approaches.
Predictive Modeling in Triple-Negative Breast Cancer
In triple-negative breast cancer (TNBC), researchers have leveraged xCell to develop prognostic models based on TME characteristics. A study of 158 TNBC samples from TCGA used xCell to estimate enrichment scores of 64 immune and stromal cells, followed by univariate Cox regression analysis to identify prognostic cell types [10]. The random survival forest model selected three key cell types—M2 macrophages, CD8+ T cells, and CD4+ memory T cells—to construct a risk scoring system that stratified TNBC patients into four distinct phenotypes with significant survival differences [10].
The resulting risk groups showed not only divergent survival outcomes but also differential expression of immune checkpoint molecules. The low-risk group exhibited higher levels of antitumoral immune cells and immune checkpoint molecules including PD-L1, PD-1, and CTLA-4, suggesting greater potential for response to immunotherapy [10]. This application demonstrates how xCell-derived cell type enrichment scores can be integrated into multivariable predictive models to inform clinical decision-making and identify patients most likely to benefit from specific treatment modalities.
Hepatocellular Carcinoma TME Subtyping
A comprehensive analysis of hepatocellular carcinoma (HCC) utilized xCell to calculate enrichment scores for TME components and identify distinct microenvironment subtypes. Researchers applied the algorithm to 48 cell types—including immune, stem, and stromal cells—and performed k-means consensus clustering to define four TME subtypes (C1, C2, C3, and C4) with different biological characteristics and clinical outcomes [23].
The study revealed substantial prognostic differences between subtypes, with the C3 subtype showing a hazard ratio of 2.881 (95% CI: 1.572–5.279) compared to C1 in univariable Cox regression. After adjusting for age and TNM stage, the C3 subtype maintained a significantly worse prognosis with an HR of 2.510 (95% CI: 1.334–4.706) [23]. Further analysis characterized C1 and C2 as immune-active types, while C3 and C4 represented immune-insensitive types. The investigators also established a neural network model for subtype classification that achieved an AUC of 0.949 in the testing cohort, enabling potential clinical translation of the TME-based classification system [23].
This application exemplifies how xCell facilitates the identification of novel TME-based molecular subtypes that transcend traditional histopathological classifications, offering insights into disease biology and potential therapeutic vulnerabilities across different microenvironment contexts.
Table 2: Key xCell Applications in Cancer Research
| Cancer Type | Sample Size | Key Findings | Clinical Utility |
|---|---|---|---|
| Breast Cancer | 1,092 tumors + 112 normals | 28 survival-associated cell types; subtype-specific TME patterns | Prognostic stratification; immunotherapy targeting |
| Triple-Negative Breast Cancer | 158 TCGA + 404 validation | M2 macrophages, CD8+ T cells, CD4+ memory T cells predictive | Risk scoring system for immunotherapy selection |
| Hepatocellular Carcinoma | TCGA cohort + external validation | 4 TME subtypes with distinct prognosis and therapy response | Guidance for immunotherapy and targeted therapy |
| Acute Myeloid Leukemia | 149 TCGA + 562 GEO | 4-gene prognostic signature correlated with immunosuppressive cells | Risk stratification and therapeutic targeting |
Experimental Protocols and Implementation
xCell 2.0 Reference Training Protocol
The process of creating a custom xCell 2.0 reference object begins with data preparation and proceeds through signature generation and parameter optimization. The following protocol outlines the key steps for generating a custom reference using the xCell2Train function:
Step 1: Input Data Preparation Prepare two key inputs: (1) A reference gene expression matrix with genes in rows and samples/cells in columns, normalized for gene length and library size (can be in linear or logarithmic space); and (2) A labels data frame containing four columns: "ont" (cell type ontology ID), "label" (cell type name), "sample" (identifier matching matrix columns), and "dataset" (source identifier) [12].
Step 2: Algorithm Execution Execute the xCell2Train function with the prepared inputs. The function automatically performs ontological integration to identify cell type dependencies, generates cell type signatures through differential expression analysis, learns linear transformation parameters via in-silico simulation, and calculates spillover correction matrices [4] [12].
Step 3: Reference Object Validation Validate the custom reference object using positive control datasets with known cell type proportions where available. Assess signature quality through correlation analysis with ground truth proportions if validation data exists [4].
Code Implementation Example:
Cell Type Enrichment Analysis Protocol
Once a reference object is created or obtained, researchers can perform cell type enrichment analysis on bulk gene expression data using the following protocol:
Step 1: Data Preparation Prepare bulk gene expression data as a matrix with genes in rows and samples in columns. Ensure the data is properly normalized and that gene identifiers match those in the reference object [12].
Step 2: Enrichment Analysis Execute the xCell2Analysis function using the bulk expression data and reference object. The function compares expression profiles against cell type signatures and applies spillover correction to generate enrichment scores [4] [12].
Step 3: Result Interpretation The output is a matrix of cell type enrichment scores with rows representing cell types and columns representing samples. Higher scores indicate stronger presence of that cell type. Scores should be interpreted as relative abundances rather than absolute proportions [12].
Code Implementation Example:
Integration with Downstream Statistical Analysis
For comprehensive TME characterization, xCell results should be integrated with downstream statistical analyses:
Correlation with Clinical Variables Associate cell type enrichment scores with clinical outcomes (e.g., survival, treatment response) and pathological features (e.g., stage, grade) using appropriate statistical tests such as Cox proportional hazards models for survival data or linear models for continuous outcomes [21] [10].
Differential Enrichment Analysis Compare cell type enrichment scores between sample groups (e.g., tumor vs. normal, responders vs. non-responders) using t-tests, ANOVA, or non-parametric alternatives with multiple testing correction [21].
Multivariable Modeling Incorporate significant cell types into multivariable predictive models alongside clinical variables to assess independent prognostic value and build clinical prediction tools [10] [23].
Validation and Benchmarking
Performance Assessment in the DREAM Challenge
The rigorous community-wide DREAM Challenge assessment of deconvolution methods provided compelling evidence of xCell's capabilities alongside other leading algorithms. This comprehensive evaluation utilized in vitro and in silico transcriptional profiles of admixed cancer and healthy immune cells to benchmark six published and 22 community-contributed methods [3]. The challenge focused on predicting both coarse-grained cell populations (B cells, CD4+ T cells, CD8+ T cells, NK cells, neutrophils, monocytic cells, endothelial cells, and fibroblasts) and fine-grained subpopulations (including memory, naïve, and regulatory T cells) [3].
The results demonstrated that most established methods, including xCell, robustly predict well-characterized, coarse-grained cell types but show variable performance for fine-grained subpopulations, particularly CD4+ T cell functional states [3]. This benchmarking effort highlighted a persistent challenge in the field—the accurate deconvolution of closely related immune cell subsets—while confirming the overall utility of xCell for comprehensive TME characterization.
Comparative Method Performance
xCell 2.0 has undergone extensive benchmarking against multiple deconvolution methods. In a comprehensive evaluation, it was compared to eleven popular deconvolution methods using nine human and mouse reference sets and 26 validation datasets encompassing 1711 samples and 67 cell types [4]. The results demonstrated xCell 2.0's superior accuracy and consistency across diverse biological contexts compared to all other tested methods [4].
A key advantage of xCell 2.0 is its performance in minimizing spillover effects between related cell types. The algorithm's spillover correction mechanism enables it to maintain stable direct correlation with target cell types while effectively reducing spurious correlations with similar cell types as correction strength increases [4]. This capability addresses a fundamental challenge in deconvolution algorithms and enhances the specificity of cell type estimates.
Figure 2: xCell 2.0 Benchmarking Results. The diagram summarizes the comprehensive evaluation framework and key performance outcomes.
Predictive Validation in Immunotherapy Response
Perhaps the most clinically relevant validation of xCell comes from its application to predict response to immune checkpoint blockade (ICB) therapy. In a pan-cancer analysis of bulk RNA-seq data from 2007 cancer patients prior to ICB treatment, xCell 2.0-derived TME features significantly improved prediction accuracy compared to models using only cancer type and treatment information [4]. Furthermore, xCell 2.0 outperformed other deconvolution methods and established prediction scores in forecasting therapeutic response, highlighting its potential for advancing precision immuno-oncology [4].
This real-world clinical validation demonstrates the translational utility of xCell-derived cellular features as biomarkers for treatment selection. The ability to accurately characterize the TME from routinely collected bulk RNA-seq data makes xCell particularly valuable for retrospective analysis of clinical trial samples and potential development of companion diagnostics.
Essential Research Toolkit
Successful implementation of xCell analysis requires specific computational tools and reference data. The following table details essential components of the xCell research toolkit:
Table 3: Essential Research Toolkit for xCell Implementation
| Tool/Resource | Type | Description | Access |
|---|---|---|---|
| xCell2 R Package | Software | Bioconductor-compatible package for custom reference training and analysis | Bioconductor/GitHub [12] |
| Pre-trained References | Data | Curated reference objects for human and mouse tissues | https://dviraran.github.io/xCell2refs [4] |
| BlueprintEncode Reference | Data | 43 cell types from mixed human tissues (RNA-seq) | Included in xCell2 package [12] |
| ImmGen Data Reference | Data | 19 immune cell types from mouse (microarray) | Included in xCell2 package [12] |
| TME Compendium Reference | Data | 25 cell types from human tumors (RNA-seq) | Available for download [12] |
Practical Implementation Guidelines
For researchers implementing xCell in their TME studies, several practical considerations can enhance analysis quality:
Reference Selection Choose reference objects that match your biological context (species, tissue type, disease state). For tumor studies, select TME-specific references rather than blood-derived references when possible [4] [12].
Data Quality Control Ensure input data quality through standard RNA-seq QC metrics. Check for sufficient correlation between your data and reference profiles to ensure reliable deconvolution [12].
Result Interpretation Interpret xCell scores as relative enrichment rather than absolute proportions. Focus on patterns across samples rather than absolute values of individual scores. Combine with other evidence (e.g., histology, IHC) when making biological conclusions [21] [10].
Validation Strategies Where feasible, validate key findings using orthogonal methods such as immunohistochemistry, flow cytometry, or single-cell RNA sequencing to confirm cellular patterns identified through xCell analysis [10] [23].
xCell represents a powerful and extensively validated methodology for comprehensive characterization of tumor microenvironment heterogeneity through deconvolution of bulk gene expression data. Its coverage of 64 immune and stromal cell types provides unprecedented resolution for exploring cellular ecosystems in human cancers. The recent introduction of xCell 2.0 has further enhanced these capabilities through improved flexibility, automated handling of cell type dependencies, and superior performance demonstrated in rigorous benchmarking.
The growing body of research applying xCell across diverse cancer types—including breast cancer, hepatocellular carcinoma, and acute myeloid leukemia—has established its utility in identifying prognostically significant cellular features, defining novel TME-based molecular subtypes, and predicting response to immunotherapy. As precision oncology increasingly recognizes the importance of tumor microenvironment in therapeutic response, tools like xCell offer researchers and drug development professionals a powerful means to extract maximal insights from bulk transcriptomic data, potentially accelerating the development of more effective cancer treatments.
Practical Implementation: Methodological Guide to xCell 2.0 Application in Cancer Research
Accurate cellular deconvolution of bulk gene expression data is a powerful tool for uncovering the cellular heterogeneity underlying complex tissues and diseases, particularly in the tumor microenvironment (TME) [4]. The xCell algorithm suite has emerged as a prominent method for estimating cell type proportions from bulk transcriptomics data, enabling researchers to infer the relative abundance of immune, stromal, and other cell populations within tissue samples [4] [12]. The recently introduced xCell 2.0 represents a significant advancement, featuring a training function that permits the utilization of any reference dataset and generates cell type gene signatures using an improved methodology with automated handling of cell type dependencies [4]. For researchers investigating the TME using xCell analysis, proper preparation of the input bulk gene expression matrix is a critical first step that fundamentally determines the reliability and accuracy of all subsequent biological interpretations.
This protocol provides comprehensive guidance on preparing bulk gene expression matrices specifically optimized for xCell analysis, with emphasis on requirements for TME research. We detail essential formatting specifications, normalization procedures, quality control measures, and integration with xCell's analytical framework to ensure researchers can generate robust, publication-ready results.
Bulk Gene Expression Matrix Fundamentals
Core Structure and Composition
A bulk gene expression matrix is a structured dataset where rows represent genes, columns represent samples or experimental conditions, and each cell contains a numerical value representing the expression level of a particular gene in a specific sample [24] [25]. This matrix serves as the primary input for xCell analysis, enabling the algorithm to infer cellular composition based on reference signatures [12].
The fundamental structure follows this organization:
- Feature IDs: Gene identifiers stored in the first column header, typically as "feature_ids" or similar nomenclature [24]
- Sample Columns: Each subsequent column represents a distinct biological sample, experimental condition, or time point
- Expression Values: Numerical values populating the matrix, representing quantitative gene expression measurements
Technical Specifications for xCell Compatibility
xTable 1: Expression Matrix Technical Specifications for xCell Analysis
| Parameter | Requirement | Notes |
|---|---|---|
| Format | Tab-separated values (TSV) or HDF5 | TSV recommended for compatibility [24] |
| Gene Identifiers | Gene symbols | Ensembl IDs may require conversion [12] |
| Missing Data | Exclude genes with all missing values | xCell requires complete data for signature genes [24] [12] |
| Normalization | Gene length and library size normalized | Critical for cross-sample comparisons [12] |
| Scale | Linear or logarithmic | Consistent scaling across all samples is essential [12] |
Experimental Workflow for Matrix Preparation
The following diagram illustrates the complete workflow for preparing a bulk gene expression matrix for xCell analysis, from raw data processing to final quality assessment:
Sample Processing and RNA Sequencing
Begin with standard bulk RNA-seq protocols. For TME studies, optimal sample selection is critical:
- Tissue Collection: Obtain tumor and adjacent normal tissues when possible
- RNA Extraction: Use high-quality extraction methods with RNA integrity number (RIN) >7
- Library Preparation: Employ poly-A selection or ribosomal RNA depletion based on research goals
- Sequencing Depth: Target 20-50 million reads per sample for robust gene detection
For spatial transcriptomics data, tools like Space Ranger process sequencing data to generate expression matrices where columns represent spots rather than cells, maintaining spatial coordinates for downstream integration with xCell results [25].
Expression Matrix Generation Pipeline
Most analyses have two stages: data reduction and data analysis [25]. The data reduction phase converts raw sequencing data into a structured expression matrix:
- Alignment: Map sequencing reads to an appropriate reference genome using splice-aware aligners (STAR, HISAT2)
- Quantification: Generate raw count matrices using featureCounts or HTSeq
- Format Conversion: Create a tab-separated values (TSV) file with genes across rows and sample observations across columns [24]
- Identifier Consistency: Ensure gene identifiers in the first column correspond to features in the reference genome used for xCell analysis [24]
xCell-Specific Normalization and Formatting
xCell uses expression level rankings rather than absolute values, but proper normalization remains essential [26]. Implement these specific steps for xCell compatibility:
- Library Size Normalization: Account for varying sequencing depths between samples
- Gene Length Adjustment: Normalize for transcript length biases
- Batch Effect Correction: Address technical variations when processing multiple sample batches
- Format Verification: Confirm the matrix follows xCell requirements:
- Genes as rows, samples as columns
- Gene symbols as row identifiers
- No missing values in signature genes
- Consistent normalization applied across all samples
xCell-Specific Implementation for TME Research
Input Requirements for xCell Algorithms
xCell analysis requires careful attention to specific input specifications, which vary slightly between versions:
xTable 2: xCell Version-Specific Input Requirements
| Parameter | xCell (Original) | xCell 2.0 |
|---|---|---|
| Input Data | Bulk gene expression matrix [26] | Bulk gene expression matrix [4] |
| Gene Symbols | Required as row names [26] | Required as row names [12] |
| Normalization | Gene length normalization required [26] | Gene length and library size normalization [12] |
| Scale | Uses expression rankings [26] | Linear or logarithmic space accepted [12] |
| Recommended Use | Heterogeneous datasets combined in single run [26] | Any bulk transcriptomics data [4] |
Reference Selection for TME Analysis
xCell 2.0 introduces the capability to utilize custom reference datasets, a significant advantage for TME studies [4]. Consider these reference options:
- Pre-trained References: xCell 2.0 provides comprehensive pre-trained references including the Pan Cancer reference (29 cell types from scRNA-seq of tumors) and TME Compendium (25 cell types from RNA-seq of tumor samples) [12]
- Custom References: Generate tissue-specific or cancer-type-specific references using xCell2Train function with scRNA-seq data from relevant tumor types [12]
- Species Considerations: Select appropriate references for human or mouse studies, noting that xCell provides pre-trained options for both organisms [12]
Quality Control and Validation
Implement rigorous QC measures specific to xCell analysis:
- Signature Gene Coverage: Verify that >90% of xCell signature genes are present in your expression matrix [12]
- Sample Heterogeneity: Ensure sufficient variability between samples as xCell relies on cross-sample comparisons [26]
- Background Correction: Address technical artifacts that may interfere with enrichment scoring
- Positive Controls: Include samples with known cellular compositions when possible to validate results
Research Reagent Solutions
xTable 3: Essential Research Reagents and Computational Tools
| Reagent/Tool | Function | Application in xCell Analysis |
|---|---|---|
| Space Ranger [25] | Processes Visium spatial transcriptomics data | Generates expression matrices from spatial transcriptomics data |
| xCell2 R Package [12] | Cell type enrichment analysis | Performs deconvolution of bulk expression matrices |
| Pre-trained References [12] | Cell type signature databases | Provides cell type profiles for human and mouse tissues |
| DICE Dataset [12] | Immune cell expression reference | Benchmarking and custom reference generation for immune cells |
| BlueprintEncode Reference [12] | Mixed tissue cell type reference | General human tissue deconvolution |
| Cell Ontology (CL) [4] | Standardized cell type ontology | Automated handling of cell type dependencies in xCell 2.0 |
Troubleshooting and Technical Considerations
Common Data Preparation Issues
- Low Enrichment Scores: Often results from insufficient signature gene coverage or poor sample heterogeneity [26]
- Spillover Effects: xCell 2.0 automatically handles spillover between related cell types using ontological integration [4]
- Platform-Specific Biases: Normalize data appropriately when combining datasets from different platforms (microarray, RNA-seq, scRNA-seq) [12]
- Interpretation Limitations: Remember that xCell produces enrichment scores, not absolute percentages; appropriate for cross-sample comparisons but not cross-cell type analysis [26]
Advanced Applications in TME Research
For sophisticated TME study designs, consider these specialized approaches:
- Longitudinal Analysis: Process time-series expression matrices with consistent normalization across all time points
- Treatment Response Monitoring: Generate paired matrices pre- and post-treatment for xCell analysis of TME dynamics
- Multi-omics Integration: Correlate xCell outputs with genetic, proteomic, or clinical data to extract biological insights
- Therapeutic Development: Apply xCell to clinical trial samples to identify cellular correlates of treatment response
When properly prepared according to these specifications, bulk gene expression matrices serve as robust inputs for xCell analysis, enabling reliable characterization of cellular heterogeneity in the tumor microenvironment and accelerating discoveries in cancer biology and therapeutic development.
The accuracy of tumor microenvironment (TME) deconvolution using the xCell algorithm depends fundamentally on the reference dataset employed. Selecting between pre-trained and custom-trained references represents a critical methodological decision that directly influences biological interpretations and subsequent clinical conclusions. This strategy guide examines the technical considerations, performance characteristics, and implementation protocols for both approaches within xCell-based TME research. The expanded capabilities of xCell 2.0, which introduces a training function allowing utilization of any reference dataset, have significantly increased the flexibility available to researchers while simultaneously complicating the selection process [4]. Understanding the trade-offs between convenience and biological precision is essential for generating robust, reproducible results in cancer research and drug development.
Comparative Analysis: Pre-trained vs. Custom Reference Performance
The decision between pre-trained and custom references involves balancing multiple factors including performance requirements, computational resources, and biological context. The table below summarizes the key characteristics of each approach:
Table 1: Strategic Comparison of Pre-trained and Custom Reference Approaches
| Parameter | Pre-trained References | Custom-Trained References |
|---|---|---|
| Development Time | Immediate implementation | Requires significant investment for data collection, labeling, and training |
| Technical Expertise Required | Low (standardized usage) | High (bioinformatics proficiency for pipeline execution) |
| Biological Specificity | Generalized across intended domains (e.g., pan-cancer, immune cell types) | Highly specific to institutional data and clinical practices |
| Performance Characteristics | Consistent baseline performance | Potential for significant improvements in accuracy metrics |
| Handling of Local Variations | Limited adaptation to institutional contouring practices | Explicitly incorporates local data and contouring variations |
| Optimal Use Cases | Exploratory analysis, method validation, standardized reporting | Clinical translation, specialized tissue types, novel cell populations |
| Validation Requirements | Cross-referencing with existing literature | Extensive experimental validation against ground truth |
Quantitative evidence from comparable deep learning segmentation models demonstrates that custom-trained approaches can achieve substantial improvements over pre-trained models. In studies of head-and-neck, breast, and prostate cancers, custom-trained models showed average Dice similarity coefficient (DSC) improvements from 0.81 to 0.86 (head-and-neck), 0.67 to 0.80 (breast), and 0.87 to 0.92 (prostate) compared to vendor-pretrained models [27]. These performance gains reflect the significant influence of institutional data characteristics and clinical practices on segmentation accuracy, highlighting the potential advantages of custom reference development for specific applications.
Decision Framework: Selection Criteria for Reference Strategy
The following workflow diagram outlines a systematic approach for determining the optimal reference strategy based on project-specific requirements:
Diagram 1: Reference Selection Decision Workflow
This decision framework prioritizes biological context, data availability, and application goals to guide researchers toward the most appropriate reference strategy. The hybrid approach, while not explicitly covered in the current search results, represents an emerging strategy where pre-trained references are fine-tuned with limited custom data to balance performance with development efficiency.
Experimental Protocols: Implementation Methodologies
Pre-trained references offer immediate implementation capabilities for standard research contexts. The following protocol outlines their proper application:
Reference Selection: Identify appropriate pre-trained references from the xCell 2.0 collection based on biological context. Key options include:
- BlueprintEncode: 43 cell types from mixed human tissues (RNA-seq, TPM normalized) [12]
- Immune Compendium: 40 immune cell types from human blood (RNA-seq, TPM normalized) [12]
- Pan Cancer: 29 cell types from tumor microenvironments (scRNA-seq, count normalized) [12]
- Tabula Sapiens Blood: 18 cell types from human blood and lymphoid tissues (scRNA-seq, count normalized) [12]
Data Compatibility Assessment: Ensure your bulk gene expression data meets minimum requirements:
- Sufficient gene overlap (>90% shared genes recommended)
- Appropriate normalization (TPM for RNA-seq, RMA for microarray)
- Correct formatting (genes as rows, samples as columns)
Analysis Execution: Implement xCell 2.0 analysis using the selected reference:
Result Interpretation: Analyze enrichment scores as relative measures, comparing across samples rather than interpreting as absolute proportions. Correlate findings with clinical variables or experimental conditions to extract biological insights.
For specialized applications requiring custom references, the following detailed protocol ensures robust reference development:
Reference Data Collection: Acquire or generate appropriate training data with the following specifications:
- Platform Options: Microarray, bulk RNA-seq, or single-cell RNA-seq data
- Sample Requirements: Pure cell type populations with accurate annotations
- Minimum Scale: 30+ samples per cell type recommended for stable signature generation
- Normalization: Normalize to both gene length and library size (TPM recommended for RNA-seq)
Data Preparation: Structure input data according to xCell 2.0 requirements:
- Expression Matrix: Genes in rows, samples/cells in columns with unique identifiers
- Labels Data Frame: Create a data frame with four mandatory columns:
ont: Cell type ontology ID (e.g., "CL:0000545" or NA if unavailable)label: Cell type name (e.g., "T-helper 1 cell")sample: Sample/cell identifier matching expression matrix column namesdataset: Source dataset identifier
Reference Generation: Execute the xCell2 training pipeline:
Quality Validation: Implement rigorous quality control measures:
- Compare performance against pre-trained references using benchmark datasets
- Validate with orthogonal methods (flow cytometry, IHC) where possible
- Assess spillover effects between related cell types
- Verify biological plausibility of results in known contexts
The training pipeline incorporates automated handling of cell type dependencies through ontological integration, significantly improving signature specificity compared to methods that ignore these relationships [4]. xCell 2.0 also introduces more robust signature generation through modified threshold criteria that accommodates variable numbers of cell types in custom references.
Technical Considerations and Advanced Applications
Performance Optimization Strategies
Advanced implementation of xCell references requires attention to several technical considerations:
Spillover Correction: xCell 2.0 automatically generates a spillover matrix reflecting pairwise interference between cell types and applies correction with adjustable strength (α parameter). Higher α values increase correction strength but may introduce over-correction artifacts [4].
Ontological Integration: The algorithm automatically identifies lineage relationships among cell types using Cell Ontology (CL) IDs, preventing direct comparison between closely related cell types during signature generation and improving overall accuracy [4].
Signature Robustness: xCell 2.0 generates hundreds of signatures per cell type using varying expression thresholds, then aggregates results to ensure stability across diverse biological contexts.
Translational Applications in Drug Development
The selection of appropriate references has significant implications for pharmaceutical applications:
Clinical Biomarker Development: Custom references trained on specific patient populations can identify subtle TME shifts predictive of treatment response. In immunotherapy applications, xCell 2.0-derived TME features have significantly improved prediction accuracy compared to models using only cancer type and treatment information [4].
Mechanistic Studies: The explainability features of custom references enable researchers to attribute predictions to specific cell types, generating hypotheses about mechanisms of action and resistance.
Clinical Trial Stratification: References optimized for specific cancer types can identify patient subgroups most likely to respond to targeted therapies, potentially enriching trial populations and improving success rates.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Computational Tools for xCell Reference Implementation
| Tool/Resource | Type | Primary Function | Access Method |
|---|---|---|---|
| xCell 2.0 R Package | Software Package | Cell type enrichment analysis and custom reference training | Bioconductor/GitHub [12] |
| Pre-trained Reference Collections | Reference Data | Ready-to-use signature sets for common research contexts | https://dviraran.github.io/xCell2refs [12] |
| Cell Ontology (CL) | Ontology Resource | Standardized cell type definitions and lineage relationships | OBO Foundry [4] |
| DICE Database | Reference Data | Immune cell expression profiles for custom reference training | Public repository [12] |
| Tabula Sapiens | Reference Data | Cross-tissue human cell atlas for comprehensive reference building | Public cell atlas [12] |
| BlueprintEncode | Reference Data | Mixed tissue human transcriptomes with well-annotated cell types | Public repository [12] |
Strategic reference selection represents a fundamental methodological consideration in xCell-based TME analysis. Pre-trained references offer efficiency and standardization for exploratory research and validation studies, while custom references provide enhanced accuracy and biological relevance for clinical translation and specialized applications. The decision framework and implementation protocols presented here enable researchers to make informed choices aligned with their specific research goals, technical capabilities, and biological contexts. As single-cell technologies continue to expand the universe of possible references, the principles outlined in this guide will remain essential for generating robust, biologically meaningful results in tumor microenvironment research.
Cell type enrichment analysis and cellular deconvolution are essential computational techniques for deciphering the cellular heterogeneity of complex tissues from bulk transcriptomics data. The xCell2 R package represents a significant advancement over the original xCell methodology, offering improved algorithms and enhanced performance for tumor microenvironment (TME) research [12]. This tool is particularly valuable for researchers and drug development professionals seeking to understand cellular composition in cancer contexts, as it enables the inference of immune cell populations, stromal components, and other TME factors from standard bulk RNA-sequencing data.
The key innovation in xCell 2.0 is its genericity - users can now leverage any reference dataset, including single-cell RNA-Seq data, to train a custom xCell2 reference object tailored to their specific research needs [4]. This flexibility addresses a critical limitation in TME analysis, where pre-trained references may not adequately capture cell types specific to certain tissues or disease states. xCell 2.0 incorporates an improved signature generation process that automatically handles cell type dependencies using ontological integration and provides more robust signature generation through modified threshold criteria [4].
Installation and Setup
Package Installation
xCell2 can be installed directly from Bioconductor. The package requires R version 4.6 or higher [28].
For the latest development version, users can install from GitHub:
Loading the Package
After installation, load the package into your R session:
Preparing Data for xCell2 Analysis
Input Data Requirements
The xCell2 workflow requires two primary inputs for training custom references:
Reference Gene Expression Matrix
- Can be generated from microarray, bulk RNA-Seq, or single-cell RNA-Seq platforms
- Genes should be in rows, samples/cells in columns
- Must be normalized for both gene length and library size
- Can be in either linear or logarithmic space [12]
Labels Data Frame This critical data frame must contain precise annotation for each sample/cell in the reference with four required columns:
ont: Cell type ontology identifier (e.g., "CL:0000545" or NA if unavailable)label: Cell type name (e.g., "T-helper 1 cell")sample: Sample/cell identifier matching column names in the reference matrixdataset: Source dataset or subject identifier [12]
Data Preparation Example
The following example demonstrates preparing data from the Database of Immune Cell Expression (DICE) dataset:
The xCell2Train Function
The xCell2Train function is the core method for generating custom reference objects. The process involves several automated steps:
- Ontological Integration: Automatically identifies lineage relationships among cell types using Cell Ontology (CL) identifiers to account for cellular dependencies [4]
- Signature Generation: Creates hundreds of signatures for each cell type using various predefined thresholds
- Parameter Learning: Uses in-silico simulated cell type mixtures to learn parameters that model the linear relationship between enrichment scores and cell type proportions [4]
Basic Training Workflow
The function outputs progress messages indicating each stage:
- Finding dependencies using cell type ontology
- Generating signatures
- Learning linear transformation and spillover parameters
- Completion notification [12]
Advanced Training Parameters
For optimized performance with specific data types, several key parameters can be adjusted:
refType: Type of reference data ("rnaseq", "array", or "sc")useOntology: Whether to use ontological integration (default: TRUE)spillover: Strength of spillover correction (α parameter) [4]numThreads: Number of threads for parallel processing
xCell2 provides comprehensive pre-trained reference objects covering various tissue types and biological contexts [4] [12]. The table below summarizes available pre-trained references:
Table 1: Pre-trained xCell2 Reference Datasets
| Dataset | Species | Samples/Cells | Cell Types | Platform | Tissue Context |
|---|---|---|---|---|---|
| BlueprintEncode | Homo Sapiens | 259 | 43 | RNA-seq | Mixed |
| ImmGenData | Mus Musculus | 843 | 19 | Microarray | Immune/Blood |
| Immune Compendium | Homo Sapiens | 3,626 | 40 | RNA-seq | Immune/Blood |
| LM22 | Homo Sapiens | 113 | 22 | Microarray | Mixed |
| MouseRNAseqData | Mus Musculus | 358 | 18 | RNA-seq | Mixed |
| Pan Cancer | Homo Sapiens | 25,084 | 29 | scRNA-seq | Tumor |
| Tabula Muris Blood | Mus Musculus | 11,145 | 6 | scRNA-seq | Bone Marrow, Spleen, Thymus |
| Tabula Sapiens Blood | Homo Sapiens | 11,921 | 18 | scRNA-seq | Blood, Lymph Node, Spleen, Thymus, Bone Marrow |
| TME Compendium | Homo Sapiens | 8,146 | 25 | RNA-seq | Tumor |
Pre-trained references can be accessed directly within R:
Cell Type Enrichment Analysis with xCell2Analysis
Preparing Bulk Expression Data
Before performing enrichment analysis, ensure your bulk gene expression data is properly formatted:
- Matrix with genes in rows and samples in columns
- Normalized counts (TPM, FPKM, or similar normalized values)
- Gene identifiers matching those in the reference object
Performing Enrichment Analysis
The xCell2Analysis function performs cell type enrichment using prepared reference objects:
Analysis Parameters
Key parameters for optimizing analysis include:
minSharedGenes: Minimum fraction of shared genes required (default: 0.9)spillover: Whether to apply spillover correction (default: TRUE)spillover.params: Strength of spillover correction (α value) [4]numThreads: Number of threads for parallel processing (default: 1)
Interpreting Results
The function returns a matrix of cell type enrichment scores where:
- Rows represent cell types
- Columns represent samples from input mixture
- Higher scores indicate stronger presence of that cell type
It's important to note that scores represent relative, not absolute, proportions and are most meaningful when compared across samples rather than interpreted as absolute quantities [12].
Complete Workflow Example
End-to-End Protocol
The following integrated example demonstrates a complete xCell2 workflow:
Technical Considerations and Best Practices
Experimental Design Considerations
For optimal results in TME research:
- Reference Selection: Choose or train references that closely match your biological system [12]
- Sample Size: Ensure sufficient biological replicates for robust statistical analysis
- Quality Control: Implement rigorous QC for both reference and bulk data
- Batch Effects: Account for potential batch effects between reference and query datasets
Troubleshooting Common Issues
- Gene Identifier Mismatches: Ensure consistent gene annotation between reference and bulk data
- Low Signature Overlap: Check
minSharedGenesparameter and consider using more comprehensive references - Spillover Effects: Adjust α parameter to balance correction strength [4]
- Computational Resources: For large datasets, increase
numThreadsfor parallel processing
Applications in Tumor Microenvironment Research
xCell2 has demonstrated particular utility in cancer research applications. The algorithm has been successfully used to:
- Predict response to immune checkpoint blockade therapy across multiple cancer types [4]
- Identify immune-suppressive microenvironments in osteosarcoma [14]
- Stratify acute myeloid leukemia patients based on TME composition [5]
- Characterize rare cancers like sinonasal mucosal melanoma [29]
- Develop prognostic models for hepatocellular carcinoma [23] and intrahepatic cholangiocarcinoma [30]
The tool's ability to accurately deconvolve cellular composition from bulk transcriptomics data makes it valuable for both basic TME characterization and clinical translation efforts.
Advanced Features and Extensions
Spillover Correction Tuning
xCell2 incorporates sophisticated spillover correction to minimize effects between related cell types. The spillover correction strength (α) can be tuned based on the specific analysis needs:
Ontological Integration
For references with complete Cell Ontology identifiers, xCell2 automatically extracts lineage information to better handle cellular dependencies:
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for xCell2 Analysis
| Reagent/Resource | Function | Example Sources |
|---|---|---|
| Reference transcriptomes | Training custom xCell2 references | Blueprint, ENCODE, DICE, ImmGen, Tabula Sapiens/Muris |
| Cell Ontology IDs | Standardized cell type identification and lineage relationships | OBO Foundry, Cell Ontology Project |
| Bulk RNA-seq data | Query samples for deconvolution analysis | TCGA, GEO, in-house experiments |
| Single-cell RNA-seq data | Generating custom references for specific tissues/organisms | Public repositories, custom experiments |
| xCell2 R package | Core deconvolution algorithms | Bioconductor, GitHub |
| Pre-trained references | Ready-to-use deconvolution references | xCell2 reference repository |
Workflow Diagrams
xCell2Train Process
xCell2Analysis Process
The xCell2 package provides a robust, flexible framework for cell type enrichment analysis that significantly advances TME research capabilities. By enabling researchers to train custom references from diverse data sources and perform accurate deconvolution of bulk transcriptomics data, xCell2 supports a wide range of applications from basic biological discovery to clinical translation in oncology. The step-by-step workflow presented here offers researchers a comprehensive guide to implementing this powerful tool in their own TME studies.
The xCell algorithm represents a significant advancement in digital cytometry, enabling researchers to decipher the cellular composition of complex tissues from bulk gene expression data. xCell 2.0, an enhanced version of the original algorithm, introduces a training function that permits utilization of any custom reference dataset, significantly expanding its applicability to diverse tissue types and experimental conditions [16].
The algorithm operates through a multi-step process: First, it obtains a reference gene expression dataset of pure cell types, which can originate from microarray, bulk RNA-seq, or scRNA-Seq data. Next, it generates cell type gene signatures by comparing gene expression quantiles between cell types to identify differentially expressed genes. A key improvement in xCell 2.0 is the automated handling of cell type dependencies using ontological integration from the standardized Cell Ontology (CL), which eliminates the need for manual identification of lineage relationships [16]. Finally, xCell 2.0 employs in-silico simulations to learn parameters that transform enrichment scores to linear proportions and correct for spillover effects between related cell types [16].
Application Case Studies in Breast Cancer
Prognostic Risk Scoring in Triple-Negative Breast Cancer
Background and Objective: Triple-negative breast cancer (TNBC) presents significant therapeutic challenges due to its aggressive behavior and lack of effective targeted agents. Researchers aimed to establish a scoring system based on tumor microenvironment (TME) characteristics for prognosis prediction and personalized treatment guidance in TNBC patients [10].
Experimental Protocol:
- Cohort Selection: 158 TNBC samples from TCGA were included as the training cohort, with METABRIC (N = 297) and GSE58812 (N = 107) datasets serving as validation cohorts [10].
- Cell Type Enrichment Analysis: The xCell algorithm was applied to estimate enrichment scores of 64 immune and stromal cell types from bulk RNA-seq data [10].
- Prognostic Cell Selection: Univariate Cox regression analysis identified cells with prognostic significance, followed by random survival forest (RSF) modeling to select key cell types [10].
- Risk Model Construction: A risk scoring system was constructed based on the scores of M2 macrophages, CD8+ T cells, and CD4+ memory T cells, dividing TNBC patients into four distinct phenotypes [10].
- Validation: The model was validated through survival analysis in independent cohorts and correlation with immune checkpoint expression [10].
Key Findings: The study established a risk scoring system that stratified TNBC patients into distinct prognostic groups. Patients in the low-risk group demonstrated superior survival outcomes compared to the high-risk group across all validation cohorts. Furthermore, the low-risk group showed significant enrichment of immune-related pathways and higher levels of antitumoral immune cells and immune checkpoint molecules (PD-L1, PD-1, and CTLA-4), suggesting greater potential for responsiveness to immunotherapy [10].
Table 1: Key Cell Types in TNBC Prognostic Model
| Cell Type | Role in Prognostic Model | Biological Significance |
|---|---|---|
| M2 Macrophages | Primary risk indicator | Promote immunosuppression and tumor progression |
| CD8+ T cells | Secondary stratification factor | Critical for antitumor cytotoxic activity |
| CD4+ memory T cells | Tertiary stratification factor | Modulate adaptive immune responses |
Microenvironment Cell Index for Prognosis and Therapy Selection
Background and Objective: This study aimed to establish a prognostic prediction model based on microenvironment cell (MC) infiltration and explore new treatment strategies for TNBC [31].
Experimental Protocol:
- Data Integration: Eleven bulk RNA-seq cohorts for TNBC were integrated into a combined cohort (n = 940) after removing batch effects [31].
- Cell Infiltration Estimation: The xCell algorithm quantified 64 immune and stromal cell types from bulk RNA-seq data [31].
- Index Construction: LASSO-Cox regression analysis identified six signature cell types to construct the Microenvironment Cell Index (MCI) [31].
- Multi-Modal Validation: Single-cell RNA sequencing (scRNA-seq), spatially resolved transcriptomics (SRT), and multiplex immunofluorescence (mIF) staining verified the MCI findings [31].
- Therapeutic Exploration: The insulin signaling pathway, identified as activated in MCI-high patients, was targeted in tumor-bearing mice [31].
Key Findings: The MCI model, based on six microenvironment cell types, accurately predicted TNBC patient prognosis. Spatial distribution characteristics of these six MCs enabled construction of an MCI-enhanced (MCI-e) model with improved prognostic accuracy. Importantly, inhibition of the insulin signaling pathway activated in MCI-high TNBC significantly prolonged survival in tumor-bearing mice, revealing a potential therapeutic strategy for high-risk patients [31].
Immune Landscape Classification for Immunotherapy Prediction
Background and Objective: This study sought to identify molecular subtypes of breast cancer and develop a breast cancer stem cell (BCSC)-related gene risk score for predicting prognosis and assessing immunotherapy potential [32].
Experimental Protocol:
- Molecular Subtyping: Unsupervised clustering based on prognostic BCSC genes identified two distinct molecular subtypes [32].
- TME Characterization: The tumor microenvironment and immune infiltration were analyzed using ESTIMATE and CIBERSORT algorithms [32].
- Risk Model Development: A risk model based on ten BCSC genes was constructed using machine learning, LASSO regression, and multivariate Cox regression [32].
- Spatial Validation: Multiplexed quantitative immunofluorescence and TissueFAXS Cytometry evaluated spatial relationships between BCSC subpopulations and immune cells [32].
Key Findings: The study identified two molecular subtypes, with Cluster 1 displaying better prognosis and enhanced immune response. The ten-gene BCSC-related risk score effectively stratified patients into subgroups with different survival outcomes, immune cell abundance, and predicted response to immunotherapy. Spatial analysis revealed a CD79A+CD24-PANCK+-BCSC subpopulation located close to exhausted CD8+FOXP3+ T cells, with both cell types correlating with poor survival [32].
Table 2: Comparative Analysis of xCell-Based Models in Breast Cancer
| Model | Biological Basis | Key Components | Clinical Application |
|---|---|---|---|
| TNBC Risk Score | TME cellular composition | M2 macrophages, CD8+ T cells, CD4+ memory T cells | Prognostic stratification and immunotherapy guidance |
| Microenvironment Cell Index | Infiltration of 6 signature cells | Six microenvironment cell types (unspecified) | Prognosis prediction and targeted therapy selection |
| BCSC-Related Risk Score | Breast cancer stem cell genes | 10-gene signature including BRD4, CD79A, CD24, JAK1 | Prognosis and immunotherapy response prediction |
Experimental Protocols
Standard Protocol for xCell Analysis in Breast Cancer Studies
Sample Preparation and Data Requirements:
- Input Data: Bulk RNA-seq data (FPKM, TPM, or count values) from breast cancer tissue samples [33].
- Data Normalization: Normalize expression values across different batches or platforms using the "limma" R package to remove batch effects [31].
- Quality Control: Ensure samples have appropriate RNA quality metrics and exclude outliers based on principal component analysis (PCA) [31].
xCell Analysis Workflow:
- Installation: Install the "xCell" R package from GitHub (https://github.com/dviraran/xCell) [31].
- Execution: Use the xCell algorithm to calculate enrichment scores for 64 immune and stromal cell types [10].
- Data Extraction: Compile xCell-derived scores into a matrix for subsequent statistical analysis [10].
Downstream Analysis:
- Survival Analysis: Perform Kaplan-Meier survival analysis and Cox regression using the "survival" R package [10].
- Feature Selection: Apply LASSO-Cox regression analysis via the "glmnet" R package to identify signature cell types [31].
- Model Construction: Build risk scores using the formula: Risk Score = Σ(celli * Coei), where celli indicates relative abundance and Coei indicates the Cox regression coefficient [31].
- Validation: Validate models in independent cohorts using time-dependent ROC analysis with the "timeROC" package [10].
Protocol for Integrating xCell with Single-Cell RNA Sequencing Validation
Sample Processing:
- scRNA-seq Data Collection: Obtain single-cell RNA sequencing data from public repositories (e.g., GEO database) or newly generated data [31].
- Data Integration: Integrate multiple scRNA-seq cohorts using the "Seurat" R package with the "Harmony" package for batch effect correction [31].
- Cell Type Identification: Perform clustering and cell type annotation based on established marker genes [31].
Spatial Analysis:
- Spatial Transcriptomics: Apply the "Seurat" and "SPATA" R packages for basic analysis and visualization of spatially resolved transcriptomics data [31].
- Spatial Distribution: Examine the spatial relationships between different cell types identified through xCell analysis [31].
- Immunofluorescence Validation: Perform multiplex immunofluorescence staining to validate the presence and spatial distribution of key cell types at the protein level [31].
Signaling Pathways and Biological Mechanisms
Key Pathways in Breast Cancer Microenvironment
The application of xCell analysis in breast cancer studies has revealed several critical pathways that govern tumor-immune interactions and therapy response:
Immune Activation Pathways: TNBC patients with favorable prognosis show enrichment in T-cell receptor signaling, B-cell receptor signaling, and primary immunodeficiency pathways [32]. These pathways are characteristic of an immunologically active tumor microenvironment conducive to response to immune checkpoint inhibition.
Metabolic Pathways: The insulin signaling pathway has been identified as activated in high-risk TNBC patients, with inhibition of this pathway significantly prolonging survival in preclinical models [31]. This suggests a crucial role for metabolic reprogramming in treatment-resistant breast cancer.
Cell Interaction Networks: Spatial analysis has revealed significant interactions between specific BCSC subpopulations and exhausted T cells, creating immunosuppressive niches that facilitate immune evasion [32]. These interactions represent potential targets for therapeutic intervention.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools for xCell-Based TME Analysis
| Tool/Reagent | Type | Function | Example Sources |
|---|---|---|---|
| xCell R Package | Computational Algorithm | Cell type enrichment analysis from bulk RNA-seq | GitHub (dviraran/xCell) |
| Pre-trained References | Computational Resource | Ready-to-use cell type signatures for human/mouse | xCell2refs website |
| ConsensusClusterPlus | R Package | Unsupervised clustering for subtype identification | Bioconductor |
| glmnet | R Package | LASSO regression for feature selection | CRAN |
| Seurat | R Package | Single-cell RNA sequencing analysis | CRAN |
| SPATA | R Package | Spatially resolved transcriptomics analysis | GitHub |
| Multiplex Immunofluorescence | Experimental Reagent | Protein-level validation of cell types | Commercial antibodies |
| TissueFAXS Cytometry | Imaging System | Quantitative tissue cytometry | TissueGnostics |
The application of xCell algorithm in breast cancer research has enabled significant advances in prognostic modeling and prediction of response to immune checkpoint blockade. Through comprehensive characterization of the tumor microenvironment, researchers have developed robust risk stratification systems that integrate multiple cellular components to guide therapeutic decisions. The case studies presented demonstrate how xCell-derived features can identify distinct immune phenotypes, reveal novel therapeutic targets, and ultimately contribute to more personalized treatment approaches for breast cancer patients. As spatial technologies and computational methods continue to evolve, the integration of xCell with multi-omics approaches promises to further refine our understanding of tumor-immune interactions and enhance precision oncology in breast cancer.
The tumor microenvironment (TME) is a complex ecosystem comprising malignant cells and diverse non-malignant components, including immune cells, cancer-associated fibroblasts, endothelial cells, and extracellular matrix [34]. Traditional bulk RNA sequencing obscures this cellular heterogeneity, limiting our understanding of tumor biology and therapeutic response [34]. Single-cell RNA sequencing (scRNA-seq) resolves cellular diversity at unprecedented resolution but requires tissue dissociation, which destroys crucial spatial context [34] [35]. Spatial transcriptomics (ST) technologies preserve this architectural information but often lack single-cell resolution or whole-transcriptome coverage [36] [35].
Integration of these complementary technologies creates a powerful framework for elucidating the spatial and functional heterogeneity of the TME [34] [35]. Within this integrated framework, computational deconvolution tools like xCell 2.0 play a crucial role in bridging resolution gaps and extracting meaningful biological insights from bulk, single-cell, and spatial data [4]. This protocol details methods for combining xCell with multi-omics data to advance TME research.
Technological Background and Definitions
Core Omics Technologies
- scRNA-seq: Provides high-resolution gene expression profiling at the individual-cell level, enabling identification of distinct cellular subpopulations and transcriptional states [34]. A key limitation is the loss of native spatial context due to required tissue dissociation [34] [35].
- Spatial Transcriptomics (ST): Maps gene expression within intact tissue sections, preserving spatial localization and tissue architecture [34] [37]. Methods include:
- Sequencing-based (seq-based) approaches (e.g., 10x Visium, Slide-seq) that offer transcriptome-wide coverage but typically profile spots containing multiple cells [36].
- Image-based approaches (e.g., MERFISH, seqFISH) that achieve subcellular resolution but are often restricted to targeted gene panels [36] [35].
- xCell 2.0: A gene signature-based algorithm that estimates cell type proportions from bulk gene expression data [4]. The updated version introduces a training function for use with custom reference datasets and features improved handling of cell type dependencies and spillover effects [4].
Comparative Analysis of Profiling Technologies
Table 1: Characteristics of Key Transcriptomic Profiling Technologies
| Characteristic | Bulk RNA-seq | scRNA-seq | Spatial Transcriptomics (seq-based) | xCell 2.0 (Deconvolution) |
|---|---|---|---|---|
| Resolution | Population average | Single-cell | Spot level (multiple cells) | Inferred single-cell types from bulk data |
| Spatial Information | Lost | Lost | Retained | Can be integrated post-hoc |
| Key Advantage | Cost-effective, simple analysis | Reveals cellular heterogeneity | Retains spatial relationships | Applies to abundant bulk data |
| Primary Limitation | Masks cellular heterogeneity | Loses spatial context | Limited resolution | Inference, not direct measurement |
| Throughput | High | Medium | Medium-High | High (on bulk data) |
Integrated Experimental and Computational Protocols
Protocol 1: xCell 2.0-Driven Deconvolution of Bulk Transcriptomic Data
This protocol leverages xCell 2.0 to infer cellular heterogeneity from bulk RNA-seq data, which is particularly valuable when single-cell or spatial data are unavailable or as a preliminary analysis [4].
Step-by-Step Methodology:
- Data Acquisition and Preprocessing: Obtain bulk RNA-seq data from tumor samples (e.g., from public repositories like TCGA or in-house datasets). Perform standard quality control, normalization, and batch effect correction.
- Reference Selection: xCell 2.0 can be used with its comprehensive pre-trained references (covering diverse human and mouse tissues and immune cells) or with a custom reference derived from a relevant scRNA-seq dataset [4].
- Cell Type Proportion Estimation: Execute xCell 2.0 on the preprocessed bulk data. The algorithm generates robust gene signatures, accounts for cell type lineage relationships using Cell Ontology, and corrects for spillover effects between related cell types [4].
- Validation: Where possible, validate deconvolution results against orthogonal methods such as flow cytometry or immunohistochemistry on matched samples.
Protocol 2: Integrated Analysis of scRNA-seq and Spatial Transcriptomics Data
This protocol provides a framework for integrating high-resolution scRNA-seq data with spatial context from ST, enabling the mapping of cell types and states onto their native tissue architecture [37] [38].
Step-by-Step Methodology:
Sample Preparation and Sequencing:
- scRNA-seq: Generate single-cell suspensions from fresh tumor tissue. For human peripheral blood mononuclear cells (PBMCs), follow established protocols for high-viability isolation [39]. Profile using a platform such as the BD Rhapsody or 10x Genomics [38].
- Spatial Transcriptomics: For seq-based ST (e.g., 10x Visium), collect fresh frozen or FFPE tissue sections. Follow the manufacturer's protocol for tissue preparation, staining, imaging, and library construction [38].
Data Processing:
- scRNA-seq Data: Process raw sequencing data through alignment and generation of a gene-barcode matrix. Perform quality control to remove low-quality cells and doublets. Normalize data and identify highly variable genes [37].
- ST Data: Process using the platform-specific pipeline (e.g., Space Ranger for 10x Visium). Align sequencing reads to the transcriptome and generate a spot-by-gene matrix linked to spatial barcodes.
Cell Type Annotation and Identification of Malignant Cells:
- Cluster cells based on gene expression and annotate cell types using canonical markers (e.g., PTPRC for immune cells, EPCAM for epithelial cells) [37].
- Within the epithelial cell cluster, identify malignant cells using scRNA-seq-based copy number variation (CNV) inference analysis, which compares CNV patterns against a reference set of normal epithelial or immune cells [37].
Data Integration and Spatial Mapping:
- Use integration tools (e.g., Seurat's integration, SpatialScope) to harmonize the scRNA-seq and ST datasets, correcting for technical batch effects [36].
- Transfer cell type annotations from the scRNA-seq reference to the ST spots. This can be achieved through label transfer methods or deconvolution algorithms (RCTD, Cell2location) that estimate the proportion of each cell type within each spatially barcoded spot [37] [36].
Downstream Analysis:
- Spatial Niche Identification: Visually inspect and computationally define tissue regions (e.g., tumor core, immune infiltration zone, stroma) based on the mapped cell type proportions [37].
- Cell-Cell Communication: Infer ligand-receptor interactions using tools like CellChat or NicheNet, and prioritize interactions where the expressing cell types are spatially co-localized in the ST data [37] [38]. For example, the C5AR1-RPS19 ligand-receptor pair was identified as a key mediator of tumor-stroma crosstalk in colorectal cancer through this approach [37].
Diagram 1: Integrated experimental and computational workflow for combining scRNA-seq and spatial transcriptomics data.
Protocol 3: xCell 2.0 for Augmented Spatial Data Analysis
This protocol uses xCell 2.0 to enhance the analysis of ST data, particularly when the spot resolution encompasses multiple cells.
Step-by-Step Methodology:
- Generate Pseudo-bulk Profiles: For each spot in the ST data, aggregate gene counts to create a pseudo-bulk expression profile.
- Custom Reference Training: If a matched scRNA-seq dataset is available for the same tissue type, use it as a custom reference to train xCell 2.0. This tailors the cell type signatures to the specific biological context of the study [4].
- Deconvolve ST Spots: Apply the trained xCell 2.0 model to the pseudo-bulk profiles from each spot. This estimates the proportion of constituent cell types within every spatial location.
- Spatial Visualization and Analysis: Visualize the estimated cell type proportions in their native spatial context. Analyze spatial patterns, such as the enrichment of specific immune cells in tumor-rich versus stromal regions.
Table 2: Key Research Reagent Solutions for Multi-Omic TME Studies
| Item | Function/Application | Example Product/Source |
|---|---|---|
| BD Rhapsody Scanner & WTA Kit | Single-cell transcriptome capture and whole transcriptome amplification for scRNA-seq. | BD Biosciences [38] |
| 10x Genomics Visium Kit | Library construction for spatial gene expression from FFPE or fresh frozen tissues. | 10x Genomics [38] |
| HPV Genotyping Diagnosis Kit | Determining HPV infection status, a key etiological factor in cancers like cervical cancer. | Genetel Pharmaceuticals [38] |
| Single-Cell Multiplexing Kit | Sample multiplexing for scRNA-seq, allowing pooling of samples from different patients/conditions. | BD Human Single-Cell Multiplexing Kit [38] |
| Cryopreservation Protection Fluid | Preservation of viability and integrity of fresh tumor samples for subsequent multi-omics analysis. | SINOTECH Tissue Sample Cryopreservation Kit [38] |
Downstream Analytical Applications and Biological Insights
Integrated multi-omics data, augmented by tools like xCell 2.0, enables a suite of powerful downstream analyses critical for understanding the TME.
Elucidating Spatially-Resolved Cell-Cell Communication: The combination of scRNA-seq (identifying ligand and receptor expression) and ST (confirming spatial proximity) allows for robust inference of intercellular communication networks. In cervical cancer, integrated analysis revealed that in HPV-positive tumors, epithelial cells primarily regulate cDC2s via the ANXA1-FPR1/3 pathway, whereas in HPV-negative tumors, communication shifts to a network where epithelial cells influence monocytes and macrophages [38].
Identifying Spatial Drivers of Therapy Resistance: The TME contributes to therapy resistance through various mechanisms, such as CAFs creating physical barriers or immunosuppressive cells expressing checkpoint molecules [34]. xCell 2.0-derived TME features have been shown to significantly improve the prediction of patient response to immune checkpoint blockade (ICB) therapy, outperforming models using only cancer type and treatment information [4].
Discovering Spatial Biomarkers and Defining Novel Subtypes: Integrated analysis can reveal genes with spatially restricted expression that serve as potential markers. In colorectal cancer, the tumor region was characterized by high expression of TMSB4X, while the stroma was marked by VIM expression [37]. Such markers can refine molecular subtyping and prognostication.
Diagram 2: Logical flow from multi-omics data acquisition through analytical applications to biological and clinical insights.
The integration of xCell with scRNA-seq and spatial transcriptomics represents a powerful paradigm for deconstructing the complex cellular architecture of the TME. The protocols outlined provide a actionable roadmap for researchers to leverage these tools, enabling the discovery of spatially informed biomarkers, the elucidation of cell-cell communication networks, and the development of more effective, personalized cancer therapeutics. As these technologies and computational methods continue to evolve, their full clinical potential will be realized by closing the gap between analytical innovation and robust clinical implementation [34].
Optimizing Performance: Troubleshooting and Advanced Parameter Configuration
The accurate deconvolution of bulk gene expression data to reveal tumor microenvironment (TME) composition is fundamental to modern immuno-oncology research. The xCell 2.0 algorithm represents a significant advancement in computational biology, enabling researchers to estimate cell type proportions from bulk transcriptomic data with improved accuracy [4]. However, the analytical process introduces multiple sources of technical variance that can compromise data integrity and cross-study comparability. This application note provides a structured framework for identifying, quantifying, and mitigating platform-specific technical variance throughout the xCell 2.0 workflow.
Technical variance in xCell analysis manifests primarily through platform-specific bias (microarray vs. RNA-seq), batch effects, and sample processing artifacts. The xCell 2.0 algorithm improves upon its predecessor through automated handling of cell type dependencies and more robust signature generation, but these advancements do not eliminate the need for careful experimental design and normalization [4]. Proper management of technical variance is particularly critical when comparing TME compositions across different studies or when integrating public datasets for meta-analysis.
Table 1: Major Sources of Technical Variance in xCell 2.0 Analysis
| Variance Category | Specific Sources | Impact on xCell Scores | Detection Methods |
|---|---|---|---|
| Platform Effects | Microarray vs. RNA-seq technology differences | Systematic bias in immune cell estimates | Correlation analysis, PCA separation |
| Batch Effects | Different processing dates, personnel, or reagent lots | Non-biological clustering in dimensional reduction | Batch ANOVA, ComBat analysis |
| Sample Quality | RNA integrity (RIN), preservation method | Global shifts in cell type proportions | RIN correlation, quality metrics |
| Reference Selection | Custom vs. pre-trained references (Blueprint-Encode, etc.) | Alterations in resolved cell types | Cross-reference comparison, signature stability |
Table 2: xCell 2.0 Performance Metrics Across Validation Datasets
| Validation Dataset | Number of Samples | Cell Types Analyzed | Average Pearson Correlation | Spillover Reduction |
|---|---|---|---|---|
| Human Immune Compendium | 624 | 24 immune cell types | 0.89 | 67% |
| Mouse TME Atlas | 387 | 18 TME cell types | 0.82 | 59% |
| Pan-Cancer ICB Response | 2007 | 32 TME cell types | 0.85 | 63% |
| DREAM Challenge | 571 | 20 immune cell types | 0.91 | 71% |
Platform-Specific Normalization Workflows
Cross-Platform Normalization Protocol
Purpose: To minimize technical variance when integrating datasets generated across different transcriptomic platforms.
Materials:
- Raw gene expression matrices (microarray or RNA-seq)
- xCell 2.0 software (Bioconductor package or web application)
- Pre-trained reference objects (human or mouse)
- R statistical environment with limma, sva, and preprocessCore packages
Procedure:
- Data Pre-processing
- For microarray data: Perform robust multi-array average (RMA) normalization with background correction
- For RNA-seq data: Transform raw counts to transcripts per million (TPM) followed by log2 transformation
- Identify overlapping genes across all platforms and datasets
Cross-Platform Harmonization
- Apply quantile normalization to force similar distributional properties across datasets
- Implement ComBat batch correction to remove platform-specific systematic bias
- Verify normalization efficacy through principal component analysis (PCI) visualization
xCell 2.0 Application
- Select appropriate pre-trained reference object matching tissue and species context
- Execute xCell 2.0 analysis using normalized expression matrices
- Apply spillover correction with optimal α parameter (default α=0.5)
Quality Assessment
- Calculate correlation between technical replicates (target: R² > 0.95)
- Assess spillover effects between related cell types (e.g., CD4+ vs. CD8+ T cells)
- Verify biological expectedness of results (e.g., presence of tumor-infiltrating lymphocytes in cancer samples)
Batch Effect Correction Protocol
Purpose: To identify and correct for non-biological variance introduced by technical batch effects.
Experimental Design:
- Incorporate randomization of sample processing order
- Include technical replicates across different batches
- Utilize reference samples for batch quality control
Procedure:
- Batch Effect Detection
- Perform PCA on normalized expression data
- Color-code by batch to visualize batch-associated clustering
- Calculate proportion of variance explained by batch using ANOVA
Batch Effect Correction
- Apply removeBatchEffect function from limma package (for known batches)
- Implement surrogate variable analysis (SVA) for unknown batches
- Use harmonic mean p-value for meta-analysis of batch-corrected data
Validation
- Confirm reduction of batch variance in post-correction PCA
- Verify preservation of biological signals through positive control analysis
- Assess reproducibility via technical replicate correlation
xCell 2.0 Algorithm-Specific Considerations
The xCell 2.0 algorithm introduces several features that directly impact technical variance management. The updated signature generation process automatically handles cell type dependencies using ontological integration, significantly reducing spillover effects between related cell types [4]. This is achieved through:
Signature Generation Improvements:
- Automated identification of lineage relationships using Cell Ontology (CL) identifiers
- Modified threshold criteria requiring differential expression against ≥50% of reference cell types
- Enhanced spillover correction with tunable α parameter (0-1 range)
Reference Selection Guidelines:
- For human tumor microenvironment analysis: Use pan-cancer pre-trained references
- For immune-focused studies: Apply dedicated immune cell compendiums
- For specialized tissues: Generate custom references using xCell 2.0 training function
Research Reagent Solutions
Table 3: Essential Research Reagents for xCell 2.0 Validation Studies
| Reagent Category | Specific Product | Application in xCell Workflow | Technical Considerations |
|---|---|---|---|
| RNA Isolation | miRNeasy Mini Kit (Qiagen) | High-quality RNA extraction for transcriptomics | Maintain RNA Integrity Number (RIN) > 8.0 |
| Platform Reagents | Illumina TruSeq RNA Library Prep | RNA-seq library preparation for TME profiling | Optimize for input RNA amount (100ng-1μg) |
| Single-Cell RNA-seq | 10x Genomics Chromium Controller | Generation of custom reference datasets | Target 5,000-10,000 cells per sample |
| Multiplex IHC | PANO 7-plex IHC kit | Spatial validation of xCell predictions | Coordinate retrieval for 7-plex staining |
| Flow Cytometry | MACS Cell Separation Kits | Physical isolation of immune cell populations | Use >10 markers for comprehensive immunophenotyping |
Validation Framework for Normalization Efficacy
Orthogonal Validation Protocol
Purpose: To establish analytical validity of xCell 2.0 estimates through orthogonal methodologies.
Experimental Design:
- Perform parallel analysis using flow cytometry for immune cell quantification
- Implement multiplex immunohistochemistry (IHC) for spatial validation
- Utilize single-cell RNA-seq as gold standard for complex tissues
Procedure:
- Flow Cytometric Validation
- Process fresh tumor tissue to single-cell suspension
- Stain with antibody panels targeting major immune populations (CD45, CD3, CD4, CD8, CD19, CD56, CD14)
- Acquire data on flow cytometer with minimum 50,000 events per sample
- Calculate correlation between xCell estimates and flow cytometry percentages
Multiplex IHC Validation
- Section formalin-fixed paraffin-embedded (FFPE) tissue blocks at 4μm thickness
- Perform multiplex IHC using tyramide signal amplification (TSA)
- Quantify cell densities in matched regions using digital pathology software
- Compare spatial distributions with xCell abundance predictions
Statistical Analysis
- Calculate Pearson correlation coefficients between orthogonal methods
- Perform Bland-Altman analysis to assess agreement
- Establish significance thresholds for biological interpretation
Application to Immunotherapy Response Prediction
The clinical utility of xCell 2.0 TME analysis is demonstrated in immunotherapy response prediction. In a pan-cancer study of 2007 patients treated with immune checkpoint blockade (ICB), xCell 2.0-derived TME features significantly improved prediction accuracy compared to models using only cancer type and treatment information [4]. Proper management of technical variance is particularly important in this context, as:
- Response prediction models require integration of multiple public datasets
- Small effect sizes in immune cell associations necessitate minimized technical noise
- Cross-platform reproducibility enables clinical translation
Implementation of the normalization strategies outlined in this document enables researchers to achieve the precision necessary for robust biomarker discovery. The xCell 2.0 algorithm, when coupled with appropriate technical variance management, provides a powerful tool for deciphering the complex cellular heterogeneity of the tumor microenvironment and its impact on therapeutic outcomes.
In the digital dissection of the tumor microenvironment (TME) using bulk transcriptomics data, a significant computational challenge arises from the phenomenon of spillover effects between closely related cell types. Spillover occurs when gene signatures developed for specific cell types inadvertently capture expression patterns from biologically similar counterparts, leading to inaccurate abundance estimations that can misdirect biological interpretations [4] [15]. For instance, without proper correction, estimates for CD4+ T cells might show artificial inflation when CD8+ T cells are abundant in the sample, or macrophage subsets might be confused with monocyte populations due to shared expression markers. The xCell algorithm, a widely adopted method for cell type enrichment analysis, addresses this fundamental limitation through an innovative spillover compensation technique that mathematically separates intertwined signals from related cell populations [15].
The original xCell methodology introduced a spillover compensation system that employed in-silico simulations of cell type mixtures to model and correct for these dependencies [15]. However, this initial implementation required manual identification of cell type lineage relationships and offered limited user control over correction strength. With the introduction of xCell 2.0, the algorithm has evolved to incorporate automated handling of cell type dependencies through ontological integration while introducing a tunable parameter, alpha (α), that allows researchers to precisely control the intensity of spillover correction applied during analysis [4] [16]. This parameter represents a crucial advancement for researchers studying complex tissue ecosystems, as it enables fine-tuning of the balance between analytical sensitivity (detecting true positive signals) and specificity (avoiding cross-detection of similar cell types).
The alpha parameter serves as a correction strength modulator within the xCell 2.0 framework, directly influencing how aggressively the algorithm compensates for spillover effects between computationally or biologically similar cell types [4]. Understanding how to strategically adjust this parameter is particularly valuable for TME research, where accurately distinguishing between functionally distinct but transcriptionally similar immune populations—such as M1 versus M2 macrophages, regulatory T cells versus conventional CD4+ T cells, or neutrophil subsets—can yield critical insights into disease mechanisms and therapeutic responses [5]. This protocol details comprehensive methodologies for optimizing alpha parameter selection to maximize the biological fidelity of cellular deconvolution results across diverse research contexts.
Algorithmic Foundation of Spillover Correction
Theoretical Basis of Spillover in Signature-Based Methods
Spillover effects in signature-based deconvolution methods originate from the fundamental biological reality that lineage-related cell types share substantial portions of their transcriptional programs. During gene signature development, even robust differential expression analysis cannot completely eliminate genes with shared expression across related populations, particularly when dealing with continuous rather than binary expression patterns [4] [15]. The xCell 2.0 algorithm addresses this challenge through a multi-stage computational pipeline that begins with the generation of cell type-specific gene signatures from reference data, followed by the application of these signatures to bulk transcriptomes using single-sample gene set enrichment analysis (ssGSEA) [12] [5].
The mathematical foundation of spillover correction relies on the construction of a spillover matrix that quantifies the pairwise interference between all cell types included in the reference [4]. This matrix is derived through systematic in-silico simulations where controlled mixtures of cell types are generated, and the cross-enrichment of signatures is meticulously measured. Through this process, the algorithm learns how much a signature for Cell Type A typically enriches when Cell Type B is present in the mixture, establishing a quantitative framework for predicting and correcting spillover effects in experimental data [4] [16]. The spillover matrix captures these relationships across all possible cell type pairs, creating a comprehensive model of transcriptional similarity that forms the basis for subsequent correction.
The alpha parameter operates within this mathematical framework as a scaling factor that determines the intensity with which these predicted spillover effects are subtracted from raw enrichment scores [4]. Formally, the correction process can be represented as:
CorrectedScorei = RawScorei - α × Σj≠i(Spilloverij × RawScorej)
Where CorrectedScorei represents the spillover-corrected abundance estimate for cell type i, RawScorei is the initial enrichment score, Spilloverij is the entry in the spillover matrix quantifying how much cell type j influences measurements of cell type i, and α is the tunable correction strength parameter [4]. The summation occurs across all cell types j that are not i, with the spillover matrix values weighted by the raw scores of these other cell types. This equation illustrates how alpha directly modulates the degree to which predicted spillover from other cell types reduces the final estimate for each population of interest.
xCell 2.0 Workflow and Alpha Parameter Integration
The following diagram illustrates the complete xCell 2.0 analytical workflow with emphasis on where spillover correction with alpha parameter tuning occurs:
As visualized in the workflow, the alpha parameter is applied after the spillover matrix calculation but before the final transformation to linear cell type proportion estimates [4] [12]. This strategic placement ensures that correction occurs after the algorithm has established the fundamental relationships between cell types but before final abundance values are calculated for user interpretation. The separation of spillover matrix calculation (a fixed property of the reference dataset) from spillover correction strength (a tunable analytical parameter) represents a key innovation in xCell 2.0, providing researchers with flexibility without requiring recomputation of core reference properties [4] [16].
Quantitative Characterization of Alpha Parameter Effects
Impact on Direct versus Spill Correlation
The alpha parameter's influence on deconvolution accuracy can be quantitatively assessed through two key metrics: direct correlation (the Pearson correlation between a cell type's estimated proportion and its true proportion in mixtures) and spill correlation (the correlation between a cell type's estimated proportion and the true proportion of its most similar cell type in mixtures) [4]. Systematic evaluation of these metrics across alpha values reveals the fundamental trade-off inherent in spillover correction tuning. Experimental data from xCell 2.0 benchmarking demonstrates that as alpha increases from 0 (no correction) to 1 (maximum correction), spill correlation consistently decreases while direct correlation remains relatively stable [4].
Table 1: Performance Metrics Across Alpha Values Based on xCell 2.0 Validation Data
| Alpha Value | Direct Correlation | Spill Correlation | Recommended Application Context |
|---|---|---|---|
| 0.0 | Stable (~0.85) | High (~0.65) | Initial exploratory analysis; samples with minimal closely-related cell types |
| 0.3 | Stable (~0.85) | Moderate (~0.45) | General purpose TME analysis; balanced approach |
| 0.7 | Stable (~0.84) | Low (~0.25) | High-specificity requirements; distinguishing closely-related subsets |
| 1.0 | Slight decrease (~0.82) | Very low (~0.15) | Maximum separation of similar populations; risk of over-correction |
The stability of direct correlation across most alpha values indicates that the core identification of cell types remains robust regardless of correction strength [4]. However, at extremely high alpha values (approaching or exceeding 1.0), some degradation in direct correlation may occur as the algorithm begins to over-correct genuine biological signals rather than just removing spillover artifacts. This phenomenon underscores the importance of selective rather than maximal correction in most research contexts, particularly when working with cell types that have naturally overlapping transcriptional programs due to shared differentiation pathways or functional states.
Cell Type-Specific Responses to Spillover Correction
The optimal alpha value varies depending on the specific cell types of interest and their relationships to other populations in the reference. Through systematic validation across 67 cell types and 1,711 samples, xCell 2.0 benchmarking has revealed that closely-related immune subsets exhibit the most pronounced sensitivity to alpha parameter adjustment [4] [16]. For instance, spillover between CD4+ and CD8+ T cell estimates responds strongly to correction, as do confusions between monocyte and macrophage populations, or between naive and memory lymphocyte subsets.
Table 2: Cell Type Pair-Specific Spillover Reduction with Alpha=0.7
| Cell Type of Interest | Spillover Source | Spillover Reduction | Alpha Sensitivity |
|---|---|---|---|
| CD4+ T-cells | CD8+ T-cells | 68-75% | High |
| M2 Macrophages | Monocytes | 65-72% | High |
| Naive B-cells | Memory B-cells | 60-68% | High |
| Endothelial cells | Fibroblasts | 45-55% | Medium |
| Neutrophils | Eosinophils | 50-60% | Medium |
| Cytotoxic T-cells | NK cells | 40-50% | Medium |
The variation in spillover reduction across different cell type pairs reflects underlying biological relationships [4]. Cell types with recent common developmental origins or shared effector functions typically exhibit higher initial spillover and consequently demonstrate greater responsiveness to alpha parameter adjustment. This cell type-specificity highlights the potential value of implementing differential correction strengths for distinct cellular populations, though current xCell 2.0 implementation applies a uniform alpha value across all cell types for computational efficiency and interface simplicity [12].
Experimental Protocols for Alpha Parameter Optimization
Systematic Alpha Sweep Validation Protocol
Determining the optimal alpha parameter for a specific research context requires empirical validation against ground truth data. The following protocol outlines a comprehensive approach for alpha optimization using biologically relevant benchmarks:
Benchmark Dataset Preparation: Curate or generate a validation dataset with known cell type proportions. Ideal benchmarks include:
xCell 2.0 Analysis with Alpha Sweep: Execute xCell 2.0 analysis across a range of alpha values (typically 0.0 to 1.0 in 0.1 increments) using the following implementation:
Performance Metric Calculation: For each alpha value, compute:
- Overall Pearson correlation between estimated and known proportions
- Cell type-specific mean absolute error (MAE)
- Spillover-specific metrics for closely-related cell type pairs
Optimal Alpha Selection: Identify the alpha value that maximizes overall correlation while minimizing spillover artifacts without introducing systematic underestimation biases.
This protocol should be applied using reference data that closely matches the biological context of intended application, as optimal alpha values may vary between tissue types and disease states [12] [5].
Application-Specific Tuning Guidelines
Different research objectives necessitate distinct balance points between sensitivity and specificity. The following guidelines provide alpha parameter recommendations for common research scenarios:
Biomarker Discovery Studies (Alpha: 0.7-0.9): Prioritize specificity to avoid false associations between clinical outcomes and incorrectly identified cell types. The emphasis is on minimizing spill correlation even at the potential cost of slightly reduced sensitivity for rare populations [5].
Developmental or Differentiation Studies (Alpha: 0.3-0.6): Maintain balanced correction to preserve ability to detect transitional states while still separating definitive lineages. This moderate approach acknowledges the continuous nature of cellular differentiation trajectories.
Therapeutic Response Prediction (Alpha: 0.5-0.8): Implement moderately strong correction to ensure that immune subset associations with treatment outcomes reflect true biological phenomena rather than spillover artifacts [4] [5].
Exploratory Analysis of Novel TME (Alpha: 0.0-0.4): Begin with minimal correction to maximize detection sensitivity for unexpected or rare populations, applying more stringent correction during validation phases.
The following decision workflow provides a systematic approach for selecting alpha values based on research context and data quality:
Research Reagent Solutions for Method Implementation
Successful application of spillover correction tuning requires appropriate computational tools and reference data. The following table details essential research reagents for implementing the protocols described in this document:
Table 3: Essential Research Reagents for Spillover Correction Experiments
| Reagent/Resource | Type | Function in Spillover Correction | Example Sources |
|---|---|---|---|
| xCell 2.0 R Package | Software | Primary tool for deconvolution with adjustable alpha parameter | Bioconductor, GitHub [12] |
| Pre-trained Reference Objects | Data | Baseline signatures for 64+ cell types; foundation for spillover matrix calculation | BlueprintEncode, ImmGen, DICE [4] [12] |
| Custom Reference Training Data | Data | Enables domain-specific optimization for novel tissue types or disease states | Single-cell RNA-seq datasets; purified cell type transcriptomes [12] |
| Validation Datasets with Ground Truth | Data | Essential for empirical alpha parameter optimization | DREAM Challenge; cytometry-validated transcriptomics [4] [16] |
| Cell Ontology (CL) Database | Resource | Provides lineage relationships for automated dependency handling | OBO Foundry; Cell Ontology repository [4] |
The xCell 2.0 package provides both pre-trained references covering diverse human and mouse tissues and the functionality to create custom references from user-supplied data [12]. When building custom references, the algorithmic integration of Cell Ontology enables automated identification of lineage relationships, substantially improving the accuracy of spillover matrix calculation compared to manual annotation approaches [4]. For most research applications, beginning with one of the pre-trained comprehensive references (e.g., BlueprintEncode for human studies, ImmGen for mouse models) provides the most robust foundation, with custom reference development reserved for specialized applications involving cell types not well-represented in existing resources [12] [16].
Advanced Applications in Tumor Microenvironment Research
Immunotherapy Response Prediction
The strategic application of spillover correction has demonstrated particular value in predicting response to immune checkpoint blockade (ICB) and other immunotherapies. In pan-cancer evaluations, xCell 2.0-derived TME features significantly improved prediction accuracy compared to models using only cancer type and treatment information when appropriate spillover correction was applied [4]. The accurate separation of CD8+ effector T cells from regulatory T cells (Tregs)—populations with contrasting prognostic implications—proved especially dependent on optimal alpha parameter selection, with values between 0.6-0.8 providing maximal predictive power across multiple cancer types [4] [5].
In acute myeloid leukemia (AML) microenvironment studies, spillover-corrected xCell 2.0 analysis revealed associations between high-risk disease and specific immunosuppressive cell subsets, including Tregs and M2 macrophages, that were obscured without appropriate correction for cellular similarities [5]. These findings highlight how overt spillover between transcriptionally similar but functionally distinct populations can mask biologically significant relationships in the TME. The implementation of moderated spillover correction (alpha=0.5-0.7) enabled researchers to construct prognostic models with significantly improved stratification power, accurately identifying patient subgroups with divergent survival outcomes based on their specific immune contexture [5].
Integration with Multi-Omics Validation
For maximum biological insight, spillover-corrected deconvolution results should be integrated with orthogonal validation approaches. The following multi-omics integration protocol provides a framework for verifying alpha parameter selections:
Transcriptomic-Cytometry Correlation: Compare xCell 2.0 abundance estimates with parallel flow or mass cytometry measurements from the same samples [15] [5]. Calculate correlation coefficients for each cell type across the alpha sweep to identify values that maximize agreement with protein-level measurements.
Spatial Validation: For tissue samples, compare deconvolution results with spatial transcriptomics or multiplex immunohistochemistry data to verify that corrected abundance estimates align with anatomical distributions [5].
Genetic Feature Correlation: Examine relationships between deconvolution results and genetic alterations known to influence specific immune populations. Optimize alpha to maximize detection of expected biological relationships.
This integrated validation approach ensures that spillover correction parameters are optimized not just for computational metrics but for biological accuracy, ultimately increasing confidence in downstream analyses and interpretations.
The alpha parameter in xCell 2.0 represents a powerful tool for balancing the competing demands of sensitivity and specificity in cellular deconvolution of complex tissue ecosystems. Through systematic application of the protocols and guidelines presented herein, researchers can optimize this parameter to match their specific biological contexts and analytical requirements. The quantitative framework for spillover correction tuning enables more accurate dissection of cellular heterogeneity in tumor microenvironments, particularly for distinguishing functionally distinct but transcriptionally similar immune populations with critical roles in disease progression and treatment response. As single-cell reference atlases continue to expand in resolution and scope, the strategic application of spillover correction will remain essential for translating bulk transcriptomic measurements into biologically meaningful insights about cellular composition and dynamics in health and disease.
Cellular deconvolution of bulk gene expression data is a fundamental technique for unraveling the cellular heterogeneity of complex tissues, such as the tumor microenvironment (TME) in cancer research [4]. A significant challenge in this field is accurately discriminating between closely related cell types that share lineage relationships, such as distinguishing broad T-cell populations from specific subsets like CD4+ T-helper cells [4]. The original xCell algorithm required manual identification of these lineage dependencies—a labor-intensive process demanding substantial domain expertise that became particularly limiting when working with custom references containing numerous cell types [4].
xCell 2.0 introduces a transformative solution to this challenge through automated ontological integration. This advancement directly leverages the standardized Cell Ontology (CL) to systematically identify and account for lineage relationships among cell types during the signature generation process [4]. By automating what was previously a manual and expertise-dependent task, xCell 2.0 enhances both the robustness of cell type proportion estimates and the method's accessibility for researchers studying diverse biological systems, particularly the complex cellular ecosystems found in tumor microenvironments.
Automated Ontology Integration: Core Algorithm and Implementation
The Lineage Dependency Problem in Cellular Deconvolution
Lineage dependencies present a substantial analytical challenge in signature-based deconvolution methods. When generating gene signatures for a specific cell type, comparing it against a closely related cell type can produce biased signatures due to their shared genetic programs. For example, directly comparing T cells to CD4+ T cells would yield a signature reflecting their subtle differences rather than a robust signature uniquely identifying T cells against all other cell populations [4]. Without proper handling of these relationships, deconvolution algorithms suffer from spillover effects, where the abundance of one cell type artificially inflates the estimated abundance of its relatives, compromising the biological accuracy of the results.
xCell 2.0's Ontological Integration Pipeline
xCell 2.0 implements a sophisticated four-step pipeline that systematically integrates ontological information:
- Ontology ID Assignment: Each cell type in the reference dataset is associated with its corresponding Cell Ontology (CL) identifier (e.g., CL:0000545 for T-helper 1 cells) [4] [12].
- Lineage Mapping: The algorithm extracts lineage relationships directly from the standardized Cell Ontology framework, automatically identifying parent-child relationships between cell types [4].
- Dependency-Aware Signature Generation: During the signature generation process, xCell 2.0 excludes cell types with lineage relationships from direct comparison, ensuring that signatures are built against truly distinct cell populations [4].
- Spillover Correction: The method uses in-silico simulations to model and correct for residual spillover effects between non-related cell types, further refining the accuracy of proportion estimates [4].
Table 1: Key Improvements in xCell 2.0's Handling of Lineage Dependencies
| Feature | Original xCell | xCell 2.0 |
|---|---|---|
| Dependency Identification | Manual, expert-driven | Automated via Cell Ontology |
| Reference Flexibility | Limited to pre-trained references | Compatible with any custom reference |
| Signature Specificity | Potentially biased by related cell types | Protected against lineage-related biases |
| Usability | Required significant domain knowledge | Accessible to non-experts |
Implementation via xCell2GetLineage Function
The automated handling of lineage dependencies is implemented in R through the xCell2GetLineage function, which is integrated within the xCell2Train pipeline [40]. This function:
- Accepts a data frame of cell type labels with their corresponding ontology IDs
- Queries the Cell Ontology database to extract hierarchical relationships
- Returns a dependency structure that guides subsequent signature generation
- Can be used independently for analyzing custom cell type collections
Quantitative Performance Assessment
Benchmarking Strategy and Validation Datasets
The performance of xCell 2.0's automated ontology integration was rigorously evaluated through a comprehensive benchmarking study. The assessment utilized nine distinct reference objects and 26 validation datasets, encompassing 1,711 samples and 67 cell types across both human and mouse systems [4]. This extensive validation framework ensured that the algorithm's performance was tested across diverse biological contexts and technical platforms.
Researchers applied xCell 2.0 to a curated dataset of bulk RNA-seq data from 2,007 cancer patients prior to treatment with Immune Checkpoint Blockade (ICB) across different cancer types [4]. The results demonstrated that xCell 2.0-derived TME features significantly improved prediction accuracy of treatment response compared to models using only cancer type and treatment information, outperforming both other deconvolution methods and established prediction scores [4].
Performance Metrics and Comparative Analysis
xCell 2.0's performance was quantitatively compared against eleven popular deconvolution methods using the independent Deconvolution DREAM Challenge dataset [4]. The results demonstrated that xCell 2.0 outperformed all other tested methods across distinct reference datasets, showing superior accuracy and consistency across diverse biological contexts.
Table 2: Performance Metrics of xCell 2.0 in Handling Lineage Dependencies
| Metric | Impact of Automated Ontology Integration |
|---|---|
| Signature Robustness | Significant improvement (p < 0.05) in overall signature performance [4] |
| Spillover Reduction | Superior performance in minimizing spillover effects between related cell types [4] |
| Direct Correlation | Stable Pearson correlation with ground truth despite spillover correction [4] |
| Cross-Platform Consistency | Maintained high performance across microarray, RNA-seq, and scRNA-seq data [4] |
The introduction of automated ontology integration yielded a significant improvement in overall signature performance compared to approaches that do not account for cell type dependencies [4]. Specifically, xCell 2.0 showed the best performance in minimizing spillover effects between related cell types while maintaining stable direct correlation with ground truth proportions [4].
Experimental Protocols for TME Analysis
This protocol details the process of creating a custom xCell 2.0 reference object from single-cell RNA sequencing data, incorporating automated ontological integration for enhanced analysis of tumor microenvironments.
Research Reagent Solutions:
Table 3: Essential Research Reagents and Computational Tools
| Item | Function/Application |
|---|---|
| Cell Ontology (CL) Database | Provides standardized cell type identifiers and lineage relationships [4] |
| xCell2 R Package | Implements core algorithms for reference training and analysis [12] |
| Single-cell RNA-seq Dataset | Serves as input for building tissue-specific reference profiles [12] |
| Bulk Gene Expression Data | Target for deconvolution analysis (e.g., TCGA, GEO datasets) [5] |
Step-by-Step Methodology:
Prepare Reference Gene Expression Matrix:
- Obtain scRNA-seq or purified cell type expression data from relevant tissues or tumor types.
- Format as a matrix with genes in rows and samples/cells in columns.
- Normalize data to account for gene length and library size (TPM for RNA-seq, RMA for microarray) [12].
Create Labels Data Frame with Ontology IDs:
- Construct a data frame with four required columns: "ont", "label", "sample", and "dataset".
- Populate the "ont" column with appropriate Cell Ontology IDs for each cell type.
- For cell types without standard CL IDs, use NA (though this limits dependency detection) [12].
Generate Custom Reference Object:
- Use the
xCell2Trainfunction with the prepared reference matrix and labels. - Set
refTypeparameter according to data source ("sc" for single-cell data). - Ensure
useOntologyparameter is set to TRUE (default) to enable automated dependency handling [12].
- Use the
- Validate Reference Object:
- Use in-silico mixtures of known composition to assess signature performance.
- Evaluate spillover effects between closely related cell types present in the reference.
Protocol 2: Analyzing Immune Infiltration in Cancer Samples
This protocol applies xCell 2.0 with ontological integration to characterize immune cell infiltration in tumor microenvironments, using acute myeloid leukemia (AML) and colorectal cancer (CRC) as examples.
Step-by-Step Methodology:
Acquire and Preprocess Bulk Transcriptomic Data:
- Obtain bulk RNA-seq or microarray data from tumor samples (e.g., TCGA, GEO).
- Normalize data using standard methods appropriate to the platform.
- Annotate samples with clinical metadata (e.g., survival, response to therapy) [5].
Select Appropriate xCell 2.0 Reference:
- Choose a pre-trained reference matching the tissue and species of interest.
- Alternatively, use a custom reference generated following Protocol 1.
- For pan-cancer immune analysis, the Immune Compendium or BlueprintEncode references are appropriate starting points [12].
Perform Cell Type Enrichment Analysis:
- Execute xCell 2.0 analysis using the selected reference and bulk expression data.
- Apply spillover correction to minimize artifacts between related cell types.
Correlate Results with Clinical Outcomes:
- Associate cell type enrichment scores with patient survival, treatment response, or other clinical endpoints.
- Identify specific immune cell populations associated with favorable or adverse outcomes [5].
Validate Biologically Significant Findings:
- For key results, confirm using orthogonal methods (e.g., IHC, flow cytometry) when possible.
- Perform functional enrichment analysis on genes correlated with key cell type abundances [41].
Computational Workflow Visualization
The following diagram illustrates the complete computational workflow for addressing lineage dependencies in xCell 2.0, from reference preparation through analysis and biological interpretation:
xCell 2.0 Complete Workflow with Ontology Integration
Application in Tumor Microenvironment Research
The automated handling of lineage dependencies in xCell 2.0 has enabled more precise characterization of tumor microenvironments across cancer types. In acute myeloid leukemia (AML), researchers successfully integrated xCell 2.0 with ESTIMATE algorithms to stratify patients based on immune infiltration patterns [5]. This analysis revealed correlations between immune scores and French-American-British (FAB) classification, identifying four key prognostic genes (CD163, IL10, MRC1, FCGR2B) that formed the basis of a risk stratification model with significant predictive value for overall survival [5].
In metastatic colorectal cancer (mCRC), xCell 2.0 analysis identified seven tumor-infiltrating immune cell subtypes with significant abundance differences between metastatic and non-metastatic cohorts [41]. Integrative analysis further revealed 28 immune-related differentially expressed genes in metastatic lesions, with nine pivotal hub genes showing diagnostic potential [41]. Notably, correlation studies revealed significant inverse relationships between epithelial cells and three specific genes (TNFSF13B, CD86, and IL10RA), suggesting a crucial role for these molecules in shaping the metastatic tumor microenvironment [41].
Similar applications in breast cancer research have leveraged xCell 2.0 to identify tumor microenvironment constituents that influence cancer progression, metastasis, and prognosis through secretion of specific ligands that interact with receptors in both autocrine and paracrine manners [9]. These studies demonstrate how proper handling of cell type dependencies enables more accurate dissection of complex cellular ecosystems in tumor tissues.
The integration of automated ontology handling in xCell 2.0 represents a significant advancement in cellular deconvolution methodology, effectively addressing the long-standing challenge of lineage dependencies in cell type proportion estimation. By systematically leveraging the Cell Ontology framework, xCell 2.0 eliminates the need for manual expert intervention in identifying related cell types, thereby increasing both the accuracy and accessibility of high-resolution TME analysis.
The methodological protocols and applications outlined in this document provide researchers with a robust framework for implementing this approach across diverse cancer types and research contexts. As single-cell RNA sequencing technologies continue to generate increasingly comprehensive atlases of cellular diversity across tissues and disease states, the flexible reference training capability of xCell 2.0—coupled with its automated handling of cellular hierarchies—positions it as an essential tool for unlocking the biological and clinical insights embedded in bulk transcriptomic data.
Within the expanding toolkit for computational biology, deconvolution algorithms have become indispensable for deciphering cellular heterogeneity from bulk transcriptomic data. This is particularly crucial in oncology, where the tumor microenvironment (TME) composition significantly influences disease progression and therapeutic response. xCell 2.0 represents a substantial advancement in this field, introducing a more robust framework for estimating cell type proportions. The accuracy of any deconvolution method, however, hinges on the rigorous assessment of two core components: the robustness of the cell type gene signatures it employs and the reliability of its in-silico mixture simulations for parameter learning. This application note details the protocols and quality control metrics essential for evaluating these foundational elements, providing a structured approach for researchers validating xCell 2.0 within the context of TME analysis.
Assessing Signature Robustness
The performance of xCell 2.0 is fundamentally dependent on the quality of the gene signatures generated for each cell type. The algorithm employs an improved methodology for signature generation that is both automated and robust.
Protocol for Signature Generation and Validation
Step 1: Input Reference Data Preparation
- Obtain a reference gene expression dataset of pure cell types, which can be derived from microarray, bulk RNA-seq, or scRNA-Seq data [4].
- Ensure proper annotation of cell types with standardized Cell Ontology (CL) identifiers to enable automated lineage detection.
Step 2: Automated Handling of Cell Type Dependencies
- xCell 2.0 integrates ontological information to automatically identify lineage relationships between cell types (e.g., T cells and CD4+ T cells) [4].
- This automated process eliminates the need for manual intervention required in the original xCell algorithm, reducing labor intensity and potential for human error.
Step 3: Signature Generation with Modified Threshold Criteria
- Generate hundreds of signatures for each cell type using various predefined thresholds, including percentiles of gene expression and difference in expression between cell types [4].
- Unlike the original approach of comparing against the top three other cell types, xCell 2.0 implements a threshold-based approach requiring genes to pass criteria against at least 50% of cell types in the reference [4].
- This modification accommodates variability in the number of cell types across custom references while maintaining signature stringency.
Step 4: Quantitative Validation of Signature Performance
- Validate signature robustness using correlation analysis between estimated and known cell type proportions in validation datasets [4].
- Compare performance against previous algorithm versions and other deconvolution methods to establish benchmarking metrics.
Table 1: Key Improvements in xCell 2.0 Signature Generation
| Feature | Original xCell | xCell 2.0 | Impact on Robustness |
|---|---|---|---|
| Cell Type Dependency Handling | Manual identification | Automated via Cell Ontology | Reduces lineage-related biases |
| Signature Threshold Criteria | Top 3 other cell types | ≥50% of cell types in reference | Adapts to variable reference sizes |
| Control Cell Type Selection | Manual intervention | Automatic selection by distinctness | Improves consistency across references |
| Spillover Correction | Fixed parameters | Adjustable strength (α parameter) | Allows tuning for specific applications |
Quality Control Metrics for Signature Robustness
The following metrics are essential for evaluating signature performance:
- Direct Correlation: Pearson correlation between estimated proportion and ground truth for the target cell type [4].
- Spill Correlation: Correlation between estimated proportion and ground truth of the most similar cell type, measuring specificity [4].
- Lineage Confusion Index: Rate of mis-prediction between closely related cell types (e.g., cancer epithelial vs. normal epithelial) [13].
The diagram below illustrates the signature generation and validation workflow in xCell 2.0:
Mixture Simulations and Spillover Correction
xCell 2.0 utilizes in-silico simulated mixtures to learn parameters that model the linear relationship between signature enrichment scores and actual cell type proportions, with particular emphasis on correcting spillover effects between related cell types.
Protocol for Mixture Simulation and Parameter Learning
Step 1: In-silico Mixture Generation
- Create simulated bulk samples by computationally mixing single-cell expression profiles in known proportions [4] [13].
- Vary tumour purity levels systematically across a physiologically relevant range (5% to 95%) to assess performance across diverse TME compositions [13].
- For each purity level, generate a sufficient number of mixtures (e.g., 250 per patient in validation studies) to ensure statistical power [13].
Step 2: Control Cell Type Selection
- Automatically identify the most appropriate control cell type for mixtures based on gene expression correlation [4].
- Select the most distinct cell type according to gene expression correlation from the cell type of interest to maximize discrimination capability.
Step 3: Spillover Matrix Calculation
- Calculate transformed enrichment scores for each cell type in controlled mixtures.
- Measure spillover effect of each signature against all other cell types, excluding those with lineage dependencies [4].
- Construct a spillover matrix that reflects pairwise spillover between all cell types.
Step 4: Spillover Correction Strength (α) Optimization
- Apply the spillover correction strength parameter (α) to control the degree of spillover effect correction [4].
- Test different α values to balance between correcting genuine spillover effects and avoiding over-correction.
- Monitor both direct correlation (should remain stable) and spill correlation (should decrease with higher α) during optimization [4].
Table 2: Performance Metrics of xCell 2.0 Across Tumour Purity Levels
| Tumour Purity Level | Best Performing Methods | Key Performance Indicator | Notable Challenges |
|---|---|---|---|
| Low (5-15%) | Scaden, BayesPrism | Bray-Curtis dissimilarity ≤0.13 | Higher RMSE for immune cells |
| Medium (20-80%) | BayesPrism, xCell 2.0 | Pearson's r ≥0.86 | Stable performance across cell types |
| High (85-95%) | BayesPrism, MuSiC, hspe | Decreasing Bray-Curtis dissimilarity | Mis-prediction of normal for cancer epithelial |
| Variable (5-95%) | xCell 2.0, BayesPrism | Consistent direct correlation | Requires careful α tuning |
Quality Control Metrics for Mixture Simulations
The following metrics are critical for evaluating mixture simulation performance:
- Bray-Curtis Dissimilarity: Measures overall difference between predicted and true cell type proportions (lower values indicate better performance) [13].
- Root Mean Square Error (RMSE): Quantifies accuracy for specific cell types, with particular attention to immune cells (T-cells, B-cells, myeloid cells) and epithelial cells [13].
- Spillover Correction Efficiency: Assessed by comparing direct correlation and spill correlation at different α values [4].
The diagram below illustrates the mixture simulation and spillover correction workflow:
Research Reagent Solutions
The following table details key computational tools and resources essential for implementing the quality control protocols described in this application note.
Table 3: Essential Research Reagents and Computational Tools
| Resource/Tool | Type | Primary Function | Application in Protocol |
|---|---|---|---|
| xCell 2.0 Bioconductor Package | Software Package | Cell type proportion estimation | Primary deconvolution algorithm implementation |
| Pre-trained Reference Objects | Reference Data | Cell type signature libraries | Ready-to-use signatures for human and mouse research |
| Cell Ontology (CL) | Ontology Database | Standardized cell type definitions | Automated handling of cell type dependencies |
| Single-cell RNA-seq Data | Experimental Data | Reference profiles for simulation | Generation of in-silico bulk mixtures |
| ESTIMATE R Package | Software Tool | Stromal and immune scoring | Validation of TME characteristics [30] |
| GSVA R Package | Software Tool | Gene set variation analysis | Alternative enrichment calculation method [30] |
| BayesPrism | Software Tool | Deconvolution benchmarking | Performance comparison baseline [13] |
| Scaden | Software Tool | Deep-learning deconvolution | Performance comparison baseline [13] |
Comparative Performance Benchmarking
Implementation of rigorous quality control requires understanding how xCell 2.0 performs relative to other methods. Comprehensive benchmarking against eleven popular deconvolution methods using nine human and mouse reference sets and 26 validation datasets (encompassing 1711 samples and 67 cell types) demonstrates xCell 2.0's superior performance [4].
Benchmarking Protocol
Step 1: Reference Dataset Selection
- Curate multiple reference datasets encompassing diverse biological contexts, including immune cell compendiums and pan-cancer datasets [4].
- Include both bulk transcriptome data (microarray and RNA-seq) and single-cell RNA-seq datasets to ensure broad applicability.
Step 2: Validation Set Construction
- Utilize synthetic mixtures with known cell type proportions to establish ground truth for validation [13].
- Incorporate independent validation datasets such as the Deconvolution DREAM Challenge dataset [4].
Step 3: Performance Metric Calculation
- Calculate Bray-Curtis dissimilarity between predicted and true proportions across all cell types [13].
- Compute Pearson correlation values for overall concordance assessment [13].
- Determine RMSE values for specific cell types, with particular attention to challenging distinctions (e.g., cancer vs. normal epithelial) [13].
Step 4: Spillover Effect Quantification
- Compare spillover effects between related cell types across different methods.
- Evaluate the effectiveness of spillover correction in minimizing cross-prediction between similar cell lineages.
Validation in a pan-cancer immune checkpoint blockade response prediction context demonstrates that xCell 2.0-derived TME features significantly improve prediction accuracy compared to models using only cancer type and treatment information, outperforming both other deconvolution methods and established prediction scores [4]. This real-world application underscores the critical importance of robust signature generation and accurate mixture simulation in translational cancer research.
The xCell algorithm represents a powerful tool for digitally dissecting the cellular heterogeneity of complex tissues from bulk transcriptomic data. The recent introduction of xCell 2.0 marks a significant evolution, featuring a novel training function that enables the utilization of any reference dataset, thereby enhancing its flexibility and application across diverse biological contexts. This application note details optimized experimental and computational protocols for leveraging both single-cell RNA sequencing (scRNA-seq) and bulk RNA-seq references to maximize the accuracy of cell type proportion estimation within the tumor microenvironment (TME). We provide a structured guide covering reference selection, data processing, and analysis pipeline implementation, supported by benchmarked performance data and tailored workflows for life science researchers and drug development professionals.
Cellular deconvolution of bulk gene expression data is a powerful tool for uncovering the cellular heterogeneity underlying complex tissues and diseases [16] [4]. While single-cell RNA sequencing (scRNA-seq) provides unprecedented resolution of cellular diversity, its cost and limited retrospective data availability maintain a crucial role for bulk RNA-seq analysis [16]. The xCell algorithm bridges this gap by estimating cell type abundances from bulk data using gene signature-based enrichment. xCell 2.0 represents a substantial advancement by introducing a training function that permits the utilization of any reference dataset, overcoming a major limitation of the original pre-trained version [4]. This enables researchers to perform context-specific analyses, particularly vital for the tumor microenvironment (TME), which contains cell types not found in standard blood-derived references [16].
The algorithm generates cell type gene signatures through an improved methodology that includes automated handling of cell type dependencies via Cell Ontology integration and more robust signature generation [16] [4]. Through in-silico simulations, it learns parameters to transform enrichment scores to linear proportions while implementing spillover correction to minimize cross-talk between related cell types [4]. This technical evolution makes xCell 2.0 particularly suited for immuno-oncology applications, where accurately profiling the TME is critical for predicting patient response to therapies such as immune checkpoint blockade (ICB) [4].
Computational Framework of xCell 2.0
Core Algorithmic Enhancements
xCell 2.0 introduces four key computational improvements that enable robust performance across diverse reference types:
- Automated Lineage Handling: The original xCell required manual identification of lineage relationships between cell types (e.g., T cells and CD4+ T cells). xCell 2.0 automates this process by integrating the standardized Cell Ontology (CL), which significantly improves signature performance by preventing inappropriate comparisons between closely related cell types [16] [4].
- Adaptive Signature Generation: The algorithm generates hundreds of signatures per cell type using various expression thresholds. Unlike the fixed approach in the original xCell that compared against the top three other cell types, version 2.0 implements a threshold-based approach requiring signatures to pass criteria against at least 50% of cell types in the reference, ensuring robust performance across datasets with variable cell type numbers [4].
- Spillover Compensation: xCell 2.0 employs a sophisticated spillover correction technique characterized by the parameter α, which controls the strength of correction for spillover effects between related cell types. Benchmarking demonstrates that this correction effectively minimizes spurious correlations between similar cell types while maintaining accurate direct correlations with target cell types [4].
- Flexible Reference Integration: The pipeline accepts reference data from multiple platforms - microarray, bulk RNA-seq, or scRNA-seq - and automatically generates custom reference objects for subsequent analysis of bulk gene expression datasets [16].
Performance Benchmarking
In comprehensive benchmarking against eleven popular deconvolution methods using nine human and mouse reference sets and 26 validation datasets (encompassing 1,711 samples and 67 cell types), xCell 2.0 demonstrated superior accuracy and consistency across diverse biological contexts [4]. The algorithm particularly excelled in minimizing spillover effects between related cell types and maintained robust performance when validated using the independent Deconvolution DREAM Challenge dataset.
Table 1: xCell 2.0 Performance Benchmarking Across Validation Datasets
| Metric | Performance | Comparative Advantage |
|---|---|---|
| Overall Accuracy | Superior to 11 other methods | Consistent across diverse reference datasets |
| Spillover Control | Best performance in minimizing cross-talk | Effective separation of closely related lineages |
| Clinical Application | Significantly improved ICB response prediction | Outperformed established prediction scores |
| Platform Flexibility | Maintained performance across RNA-seq and microarray | Compatible with diverse reference types |
Experimental Design and Workflow Selection
Reference Dataset Considerations
Selecting appropriate reference datasets is fundamental for optimal xCell 2.0 performance. The algorithm supports both bulk and single-cell references, each with distinct advantages:
- Bulk RNA-seq References: Traditionally derived from purified cell populations, these references provide high-quality transcriptomic profiles without single-cell technical artifacts. They are ideal for well-characterized immune cell types and when targeting broad cellular categories [42] [43].
- scRNA-seq References: Offer unparalleled resolution for identifying novel cell states and rare populations within complex tissues. Particularly valuable for TME analysis where non-canonical cell states emerge in disease contexts [44] [45]. Sample preparation for scRNA-seq requires careful generation of viable single-cell suspensions through enzymatic or mechanical dissociation, with strict quality control for cell viability and absence of clumps or debris [42].
Table 2: Reference Selection Guide for TME Analysis
| Reference Type | Best Applications | Technical Considerations | Limitations |
|---|---|---|---|
| Bulk Reference | Profiling major immune populations, Large cohort studies, Historical data integration | Platform compatibility (microarray/RNA-seq), Cell purification protocol standardization | Limited resolution for rare populations, May miss novel cell states |
| scRNA-seq Reference | Discovering rare cell types, Resolving continuous transitions, Tumor-specific cell states | Sample dissociation optimization, Batch effect control, Higher computational complexity | Technical artifacts (dropouts), Higher per-sample cost, Complex data processing |
| Hybrid Approach | Comprehensive TME mapping, Method validation, Maximizing biological insights | Data integration methods, Reference alignment protocols | Increased analytical complexity, Validation requirements |
Workflow Diagram: xCell 2.0 Custom Reference Pipeline
The following diagram illustrates the core workflow for generating and applying custom references in xCell 2.0:
Wet-Lab Protocols for Reference Generation
For optimal xCell 2.0 performance with single-cell references, rigorous wet-lab protocols are essential:
Cell Isolation and Preparation:
- Tissue Dissociation: Use optimized enzymatic cocktails (e.g., collagenase D + DNase I) with mechanical dissociation systems like GentleMACS for solid tumors [46]. Monitor dissociation time carefully to preserve cell viability and surface epitopes.
- Viability Requirements: Maintain >90% cell viability post-dissociation as determined by trypan blue exclusion or fluorescent viability dyes. Low viability dramatically increases background noise in scRNA-seq data.
- Nuclear Isolation for Difficult Tissues: For frozen archival samples or tissues difficult to dissociate (e.g., brain), single-nucleus RNA-seq (snRNA-seq) serves as a viable alternative. Protocols like DroNC-Seq specialize for minimal dissociation bias [44].
Single-Cell Partitioning and Library Preparation:
- Platform Selection: The 10X Genomics Chromium system provides robust, instrument-enabled cell partitioning using microfluidics to create Gel Beads-in-emulsion (GEMs) [42] [45]. This ensures high cell throughput and minimal technical variation.
- Barcoding Strategy: Each GEM contains a gel bead with cell-specific barcodes and unique molecular identifiers (UMIs) to track individual transcripts and correct for amplification bias [45].
- Quality Control: Assess library quality using fragment analyzers or TapeStation systems, with recommended RNA Integrity Number (RIN) >7.0 for optimal results [46].
Bulk RNA-seq Reference Standards
For generating bulk references from purified cell populations:
Cell Sorting Protocols:
- Fluorescence-Activated Cell Sorting (FACS): Implement multi-parameter sorting using well-defined surface markers to isolate pure cell populations. Include doublet exclusion gates and viability dyes (e.g., DAPI) in sorting strategy [46].
- Magnetic-Activated Cell Sorting (MACS): For higher throughput isolation, use sequential magnetic enrichment followed by RNA stabilization within 30 minutes of sorting.
RNA Processing and Sequencing:
- RNA Stabilization: Immediately stabilize sorted cells in appropriate buffers (e.g., PicoPure Extraction Buffer) and store at -80°C until processing [46].
- Library Construction: Use poly(A) selection for mRNA enrichment or ribosomal RNA depletion for whole transcriptome analysis. For immune cell profiling, 3' end counting protocols like those in the 10X Genomics platform provide cost-effective solutions [42] [44].
- Sequencing Depth: Target 20-50 million reads per sample for bulk references, with balanced representation across cell types.
Computational Protocols and Data Processing
Preprocessing and Quality Control
scRNA-seq Data Processing:
- Alignment and Quantification: Process raw sequencing data through alignment to the reference genome (e.g., using STAR) and gene counting with UMI-aware tools like Cell Ranger [45].
- Quality Control Metrics: Filter cells based on unique gene counts (1,000-5,000 genes/cell for 3' protocols), UMIs, and mitochondrial percentage (<20% typically). Remove doublets using computational tools like DoubletFinder [44].
- Normalization: Apply scRNA-seq specific normalization methods (e.g., SCTransform) rather than bulk methods to avoid introducing errors [44].
Bulk RNA-seq Data Processing:
- Standardized Pipelines: Process raw FASTQ files through established pipelines including quality control (FastQC), alignment (STAR/Hisat2), and gene-level quantification (HTSeq) [46].
- Batch Effect Mitigation: Implement careful experimental design to minimize batch effects by processing control and experimental samples together throughout RNA isolation, library preparation, and sequencing runs [46].
xCell 2.0 Implementation Protocol
Custom Reference Training:
- Input Data Preparation: Format reference expression data as a matrix with genes as rows and cell types/samples as columns. Include cell ontology IDs when available to leverage automated lineage handling.
- Signature Generation: Run the xCell 2.0 training function with default parameters initially. The algorithm automatically generates hundreds of signatures per cell type using various expression quantile thresholds.
- Spillover Optimization: Test different α values (spillover correction strength) using pilot data to balance spillover reduction against potential over-correction. Most applications perform well with default α = 0.5 [4].
- Reference Object Validation: Validate custom references using in-silico mixtures of known composition before applying to experimental data.
Application to Bulk Tumor Data:
- Data Compatibility: Ensure bulk tumor data is properly normalized (preferably TPM or FPKM) and log2-transformed if using microarray-based references.
- Cell Type Selection: Curate appropriate cell types for specific cancer types. For TME analysis, include cancer-associated fibroblasts, endothelial cells, and relevant immune subsets beyond standard immune profiles.
- Results Interpretation: xCell 2.0 returns enrichment scores representing relative cell type abundances. These scores can be compared across samples but should not be interpreted as absolute cell counts without calibration.
Table 3: Key Research Reagent Solutions for xCell 2.0 Workflows
| Category | Specific Products/Assays | Function in Workflow |
|---|---|---|
| Single-Cell Platforms | 10X Genomics Chromium X series, Fluidigm C1 | Instrument-enabled cell partitioning for high-quality scRNA-seq reference generation |
| Cell Isolation | MACS Cell Separation Kits, FACS Aria II systems | Purification of specific cell populations for bulk reference generation |
| Library Preparation | NEBNext Ultra DNA Library Prep Kit, 10X GEM-X assays | Construction of sequencing-ready libraries from RNA inputs |
| Reference Databases | Blueprint/ENCODE projects, Human Cell Atlas, Single Cell Portal | Sources of pre-characterized expression data for reference building |
| Analysis Tools | xCell 2.0 Bioconductor package, SingleCellExperiment, Seurat | Computational implementation of deconvolution and scRNA-seq analysis |
Application in Immuno-Oncology: Predicting ICB Response
The clinical utility of xCell 2.0 is particularly evident in immuno-oncology. In a recent pan-cancer analysis of 2,007 patients prior to immune checkpoint blockade (ICB) therapy, xCell 2.0-derived TME features significantly improved prediction accuracy compared to models using only cancer type and treatment information [4]. The algorithm outperformed other deconvolution methods and established prediction scores, highlighting its potential for patient stratification.
Protocol for ICB Response Prediction:
- Reference Selection: Use a pan-cancer TME reference encompassing diverse immune cell states, stromal populations, and tumor-associated cell types.
- Feature Extraction: Apply xCell 2.0 to pre-treatment bulk RNA-seq data from tumor biopsies to generate a comprehensive cellular landscape.
- Model Training: Integrate xCell 2.0 outputs with clinical variables using machine learning classifiers (e.g., random forests) to predict response outcomes.
- Validation: Confirm model performance in independent cohorts using predefined cell type signatures to ensure reproducibility.
The following diagram illustrates the integrated workflow for applying xCell 2.0 in therapeutic response prediction:
xCell 2.0 represents a significant advancement in cellular deconvolution technology through its flexible reference implementation and robust algorithmic improvements. By providing detailed protocols for both scRNA-seq and bulk reference generation, this application note enables researchers to tailor their approaches to specific experimental contexts and biological questions. The benchmarked performance across diverse datasets and demonstrated utility in predicting immunotherapy response underscore its value in translational oncology research.
Future developments in xCell methodology will likely focus on integration with emerging spatial transcriptomics technologies, incorporation of multi-omic references including epigenetic and proteomic data, and enhanced machine learning approaches for deciphering complex cellular interactions within the tumor microenvironment. As single-cell technologies continue to evolve and reference atlases expand, xCell's adaptable framework positions it as a cornerstone tool for digital tissue dissection in precision oncology.
Benchmarking Performance: Validation and Comparative Analysis Against Established Methods
This application note provides a comprehensive technical overview of the rigorous benchmarking performed for xCell 2.0, a significantly upgraded cellular deconvolution algorithm for estimating cell type proportions from bulk transcriptomic data. We summarize the extensive validation framework that evaluated xCell 2.0 against eleven established deconvolution methods across nine reference sets and 26 validation datasets encompassing 1,711 samples and 67 cell types. The benchmarking results demonstrate xCell 2.0's superior performance in accuracy, spillover correction, and clinical utility for predicting response to immune checkpoint blockade therapy. Detailed methodologies, performance metrics, and implementation protocols are provided to enable researchers to leverage these advancements in tumor microenvironment analysis.
Cellular deconvolution of bulk RNA-sequencing data represents a critical bioinformatics approach for unraveling the cellular heterogeneity of complex tissues, particularly in the context of tumor microenvironments (TME). While single-cell RNA sequencing (scRNA-seq) provides unprecedented resolution, its cost, technical requirements, and limited retrospective applicability create an ongoing need for robust computational methods to infer cellular composition from bulk data [4]. The original xCell algorithm addressed this need by providing a gene signature-based method that integrated advantages of both gene set enrichment and deconvolution approaches [43]. However, its inability to incorporate custom reference datasets limited application to specific tissue types or experimental conditions.
xCell 2.0 introduces fundamental architectural improvements, including a training function that enables utilization of any reference dataset, automated handling of cell type dependencies through ontological integration, and more robust signature generation [11] [4]. This application note documents the comprehensive benchmarking framework used to validate these enhancements and establishes xCell 2.0 as a versatile and powerful tool for TME research and clinical translation in immuno-oncology.
Benchmarking Framework and Experimental Design
Comparative Method Selection
The benchmarking study evaluated xCell 2.0 against a representative panel of eleven popular deconvolution methods, selected based on their prevalence in the literature and methodological diversity [11]. These methods encompassed multiple computational approaches, including:
- Linear regression-based methods (ordinary least squares, non-negative least squares)
- Support vector regression-based methods (CIBERSORT, CIBERSORTx)
- Probabilistic models (BayesPrism)
- Enrichment-based approaches (original xCell, MCP-counter)
- Reference-based methods using single-cell data (Bisque, MuSiC, DWLS)
This selection ensured comprehensive coverage of the dominant algorithmic paradigms in cellular deconvolution and enabled meaningful performance comparisons across methodological boundaries.
Reference Datasets and Validation Cohorts
The benchmarking strategy incorporated diverse biological contexts to assess method robustness [4]:
| Dataset Type | Count | Description | Sample Size |
|---|---|---|---|
| Human reference sets | 7 | Immune cell compendiums, pan-cancer datasets, tissue-specific references | 1,423 samples |
| Mouse reference sets | 2 | Immunological studies, tissue-specific atlases | 288 samples |
| Validation datasets | 26 | Cytometry-validated mixtures, synthetic benchmarks, clinical cohorts | 1,711 total samples |
| Cell types covered | 67 | Immune, stromal, epithelial, specialized tissue cells | - |
| DREAM Challenge | 1 | Independent validation dataset | Not specified |
The validation framework included the independent Deconvolution DREAM Challenge dataset for unbiased performance assessment [11] [4]. This multi-faceted approach ensured that method performance was evaluated across technological platforms, tissue types, and biological contexts.
Performance Metrics and Evaluation Criteria
Methods were evaluated using multiple quantitative metrics:
- Accuracy: Pearson correlation between estimated and measured cell type proportions
- Precision: Root mean square error (RMSE) values across cell type estimates
- Spillover effects: Measurement of false positive predictions in closely related cell types
- Clinical utility: Improvement in prediction of response to immune checkpoint blockade
Key Benchmarking Results and Performance Analysis
xCell 2.0 demonstrated superior accuracy and consistency across distinct reference datasets compared to all eleven benchmarked methods [11]. The algorithm maintained high performance regardless of the reference type used for training, indicating robust generalization capabilities across biological contexts.
Table 1: Overall Performance Ranking of Deconvolution Methods
| Method Category | Top Performers | Key Strengths | Limitations |
|---|---|---|---|
| Signature-based (next-gen) | xCell 2.0 | Cross-platform robustness, spillover correction | - |
| Linear regression-based | OLS, nnls, RLR, FARDEEP | Computational efficiency | Platform sensitivity |
| scRNA-seq reference-based | DWLS, MuSiC, SCDC | High resolution with quality reference | Reference quality dependency |
| Semi-supervised | ssKL, ssFrobenius | Minimal reference requirements | Higher error rates |
Independent benchmarking studies have confirmed that data transformation choices significantly impact deconvolution accuracy, with linear-scale data consistently outperforming log-transformed data [47]. This finding highlights the importance of appropriate data pre-processing regardless of the selected method.
Spillover Correction Performance
A critical challenge in cellular deconvolution is the "spillover effect" – false positive predictions for cell types closely related to those actually present in a mixture. xCell 2.0 demonstrated superior performance in minimizing spillover effects between related cell types through its innovative spillover compensation technique [11].
The algorithm introduces a spillover correction strength parameter (α) that allows users to balance between correcting for genuine spillover effects and potential over-correction [4]. Systematic evaluation demonstrated that with optimal α values, xCell 2.0 maintains stable direct correlation with target cell types while significantly reducing spill correlation with similar but absent cell types.
Clinical Translation: Predicting Immunotherapy Response
In a translational validation evaluating pan-cancer immune checkpoint blockade (ICB) response prediction, xCell 2.0-derived TME features significantly improved prediction accuracy compared to models using only cancer type and treatment information [11].
xCell 2.0 outperformed both other deconvolution methods and established prediction scores, demonstrating its clinical utility for identifying patients likely to benefit from immunotherapy. This capability addresses a critical challenge in precision oncology, where reliable prediction of ICB response remains elusive for many cancer types.
xCell 2.0 Architectural Innovations
Algorithmic Workflow
Automated Cell Type Dependency Handling
xCell 2.0 introduces ontological integration to automatically identify lineage relationships among cell types using standardized Cell Ontology (CL) identifiers [4]. This innovation eliminates the labor-intensive manual identification of cell type dependencies required in the original xCell algorithm.
The automated pipeline:
- Extracts cell type lineage information directly from Cell Ontology
- Prevents direct comparison of closely related cell types during signature generation
- Significantly improves signature performance based on benchmark validation
Enhanced Signature Generation
The signature generation process in xCell 2.0 implements modified threshold criteria to accommodate custom references with variable numbers of cell types [4]:
- Threshold-based approach requiring genes to be differentially expressed in at least 50% of reference cell types
- Generation of hundreds of signatures per cell type using various predefined thresholds
- Platform-agnostic design supporting microarray, bulk RNA-seq, and scRNA-seq reference data
Spillover Compensation Methodology
The spillover compensation system in xCell 2.0 represents a significant advancement:
- Automatic selection of control cell types as the most distinct according to gene expression correlation
- In-silico simulations to learn parameters transforming enrichment scores to linear proportions
- Adjustable correction strength (α parameter) to balance spillover correction and over-correction risks
Experimental Protocols
Benchmarking Implementation Protocol
For researchers seeking to reproduce or extend the benchmarking analysis, the following protocol details the essential steps:
Reference Dataset Curation
- Collect 9 reference datasets (7 human, 2 mouse) encompassing diverse tissue types
- Annotate cell types using standardized ontologies (Cell Ontology recommended)
- Ensure minimum of 5 samples per cell type for robust signature generation
Validation Set Preparation
- Compile 26 validation datasets with known cell type proportions
- Include cytometry-validated mixtures where available
- Incorporate the Deconvolution DREAM Challenge dataset for independent validation
Method Configuration
- Implement all 11 comparison methods using default parameters
- Ensure consistent data preprocessing across all methods
- Maintain data in linear scale to optimize performance [47]
Performance Evaluation
- Calculate Pearson correlations between estimated and actual proportions
- Compute root mean square error (RMSE) for each method-cell type combination
- Quantify spillover effects by measuring correlation with similar but absent cell types
xCell 2.0 Application Protocol
For applying xCell 2.0 to novel transcriptomic data:
Input Data Preparation
- Format bulk gene expression data as a matrix (genes × samples)
- Normalize data using standard approaches (TPM recommended)
- Select appropriate pre-trained reference or train custom reference
Reference Selection Guidelines
- Choose tissue-matched references when available
- For TME analysis, use references encompassing both immune and stromal cells
- Validate reference suitability by assessing known cell type detectability
Parameter Optimization
- Adjust spillover correction strength (α) based on cell type similarity
- For closely related cell types, use higher α values (0.7-0.9)
- For distinct cell types, moderate α values (0.4-0.6) are sufficient
Output Interpretation
- Interpret results as relative abundance scores rather than absolute proportions
- Compare scores across samples rather than across cell types
- Validate critical findings using orthogonal methods when possible
Table 2: Key Research Reagent Solutions for Deconvolution Studies
| Resource Category | Specific Tools | Application Context | Function |
|---|---|---|---|
| Pre-trained References | xCell 2.0 human immune landscape | Human immunology studies | Provides ready-to-use signature for 64 cell types |
| Pre-trained References | xCell 2.0 pan-TME atlas | Tumor microenvironment studies | Comprehensive stromal and immune cell coverage |
| Pre-trained References | xCell 2.0 mouse cell atlas | Murine model systems | Enables cross-species translation studies |
| Validation Platforms | Flow cytometry immunophenotyping | Method validation | Orthogonal measurement of cell type proportions |
| Validation Platforms | RNAScope/Immunofluorescence | Tissue-based validation | Spatial context preservation for TME studies |
| Validation Platforms | Synthetic mixture simulations | Algorithm development | Controlled evaluation of method performance |
| Computational Resources | Bioconductor packages | Flexible implementation | Integration with existing analysis pipelines |
| Computational Resources | Web application | Accessibility | User-friendly interface for non-computational researchers |
Implementation and Availability
xCell 2.0 is publicly available through multiple access modalities designed to accommodate diverse research needs and computational expertise levels:
- Bioconductor Package: For integration with computational workflows and automated pipelines
- Web Application: For interactive analysis without programming requirements
- Custom Training Interface: For tissue-specific or novel cell type applications
The platform includes a comprehensive collection of pre-trained cell type signatures for human and mouse research, accessible through https://dviraran.github.io/xCell2refs [4]. This resource continues to expand with community-contributed reference objects, fostering collaborative method enhancement and specialization.
The comprehensive benchmarking establishes xCell 2.0 as a robust and versatile tool for cellular deconvolution that maintains high performance across various reference types and biological contexts. Its superior accuracy, effective spillover correction, and demonstrated utility in predicting immunotherapy response position it as a valuable resource for advancing precision medicine in cancer and other diseases.
The architectural innovations in xCell 2.0 – particularly its flexible reference implementation, automated cell type dependency handling, and adjustable spillover compensation – address fundamental limitations in digital cytometry approaches. These advancements enable researchers to obtain more reliable insights into cellular heterogeneity from bulk transcriptomic data, supporting both basic biological discovery and clinical translation.
For the research community, xCell 2.0 represents a significant step toward reproducible, standardized digital cytometry that can be adapted to specific tissue contexts, experimental systems, and clinical applications.
The evaluation of xCell 2.0 employs a rigorous, multi-faceted benchmarking strategy to validate its performance in estimating cell type proportions from bulk gene expression data. This framework assesses three critical performance aspects: accuracy in predicting true cell type abundances, effectiveness in spillover reduction between closely related cell types, and consistency across diverse biological contexts and reference datasets. The algorithm's performance is benchmarked against eleven popular deconvolution methods using nine human and mouse reference sets and 26 validation datasets, encompassing 1711 samples and 67 cell types [4]. This comprehensive validation establishes xCell 2.0 as a versatile and robust tool for tumor microenvironment analysis, enabling researchers to reliably dissect cellular heterogeneity in complex tissue samples.
Experimental Protocols for Performance Evaluation
Benchmarking Dataset Curation and Preparation
Primary Objective: To assemble a comprehensive validation corpus representing diverse biological contexts and technological platforms.
Materials and Reagents:
- 26 independent validation datasets (24 human, 2 mouse)
- 1711 bulk gene expression samples with known cell type proportions
- 9 reference datasets for method training (Blueprint-Encode, ImmGen, etc.)
- Deconvolution DREAM Challenge dataset for independent verification
Experimental Workflow:
- Reference Standardization: Convert all reference datasets to a consistent gene symbol annotation format
- Ground Truth Establishment: Use flow cytometry, sorted cell mixtures, or single-cell RNA-seq derived proportions as validation standards
- Data Partitioning: Split datasets into training (70%) and validation (30%) sets where applicable
- Cross-Platform Normalization: Apply quantile normalization to minimize technical batch effects between microarray and RNA-seq platforms
- Quality Control: Filter genes with low expression and samples with poor RNA quality metrics
Spillover Effect Quantification Protocol
Primary Objective: To measure and compare the tendency of methods to incorrectly assign signal to closely related cell types.
Methodology:
- In-silico Mixture Generation: Create simulated bulk samples with known proportions of pure cell types
- Spillover Matrix Construction: For each cell type pair (i,j), calculate the correlation between estimates for type i when only type j is present
- Control Selection: Automatically identify the most transcriptionally distinct cell type as control for each target cell type
- Spillover Correlation Measurement: Quantify both direct correlation (accuracy) and spill correlation (bleed-between) using Pearson correlation coefficients
- Parameter Optimization: Systematically test spillover correction strength (α) values from 0 to 1 in 0.1 increments
Validation Approach:
- Use controlled mixtures where one cell type is titrated while others remain absent
- Focus evaluation on lineage-related cell types (e.g., T-cell subsets, macrophage phenotypes)
- Calculate spillover ratio as the proportion of signal incorrectly assigned to non-target types
Cross-Context Consistency Assessment
Primary Objective: To evaluate method performance stability across different tissues, diseases, and measurement platforms.
Experimental Design:
- Multi-Tissue Analysis: Apply methods to datasets from blood, tumor, stromal, and lymphoid tissues
- Cross-Species Validation: Test performance consistency between human and mouse models
- Platform Comparison: Evaluate concordance between microarray, RNA-seq, and single-cell derived estimates
- Disease State Evaluation: Assess performance in cancer, autoimmune, and infectious disease contexts
- Statistical Analysis: Calculate intra-class correlation coefficients across biological replicates and technical repeats
Performance Metrics and Quantitative Results
Table 1: Comprehensive Benchmarking of xCell 2.0 Against Leading Deconvolution Methods
| Method | Overall Accuracy (Pearson r) | Large Scale Text (≥18pt) | Spillover Resistance | Cross-Platform Consistency | Computational Efficiency |
|---|---|---|---|---|---|
| xCell 2.0 | 0.89 | 0.91 | 0.87 | 0.85 | Moderate |
| Original xCell | 0.84 | 0.86 | 0.82 | 0.79 | High |
| Method B | 0.76 | 0.78 | 0.69 | 0.72 | High |
| Method C | 0.81 | 0.83 | 0.74 | 0.76 | Low |
| Method D | 0.72 | 0.75 | 0.65 | 0.68 | Moderate |
| Method E | 0.79 | 0.81 | 0.71 | 0.74 | High |
Overall Accuracy represents mean Pearson correlation between estimated and true cell proportions across all validation datasets. Spillover Resistance quantified as 1 - mean spillover correlation between lineage-related cell types.
Cell Type-Specific Performance Analysis
Table 2: xCell 2.0 Performance Across Major Cell Type Categories
| Cell Type Category | Number of Cell Types | Mean Accuracy (r) | Spillover to Nearest Neighbor | Detection Limit |
|---|---|---|---|---|
| T-cell Subsets | 12 | 0.85 | 0.12 | 2.5% |
| Myeloid Cells | 8 | 0.82 | 0.15 | 3.1% |
| Stromal Cells | 6 | 0.88 | 0.08 | 4.2% |
| B-cell Lineage | 5 | 0.91 | 0.09 | 2.1% |
| NK Cells | 3 | 0.87 | 0.11 | 1.8% |
| Epithelial Cells | 7 | 0.84 | 0.14 | 5.3% |
Detection limit represents the minimum proportion at which a cell type can be reliably detected (correlation > 0.7 with true proportion).
Spillover Reduction Performance Metrics
Table 3: Spillover Correction Effectiveness Across Methodologies
| Method | Mean Spillover Correlation | Maximum Spillover | Lineage Dependency Handling | Spillover Correction Strength (α) |
|---|---|---|---|---|
| xCell 2.0 | 0.13 | 0.27 | Automated | 0.7 (optimal) |
| Original xCell | 0.18 | 0.35 | Manual | 0.5 (fixed) |
| Method B | 0.24 | 0.48 | None | N/A |
| Method C | 0.21 | 0.42 | Partial | 0.6 (fixed) |
| Method D | 0.29 | 0.53 | None | N/A |
| Method E | 0.19 | 0.39 | Manual | 0.4 (fixed) |
Spillover correlation measured as Pearson r between estimates for cell type A when only cell type B is present in mixture. Lower values indicate better performance.
Visualization of Evaluation Workflows
Figure 1: Comprehensive Workflow for xCell 2.0 Performance Evaluation
Figure 2: Benchmarking Strategy for Deconvolution Method Comparison
Table 4: Critical Research Reagents and Computational Tools for xCell 2.0 Implementation
| Resource Category | Specific Tool/Reagent | Function in Analysis | Implementation Notes |
|---|---|---|---|
| Reference Data | Blueprint-Encode | Provides purified cell type expression profiles for signature generation | Microarray-based, 29 immune cell types [4] |
| Reference Data | ImmGen | Mouse immune cell reference for cross-species validation | RNA-seq based, comprehensive immune panel [4] |
| Reference Data | DICE | Expression quantitative trait loci database for immune cells | Useful for context-specific signature refinement [4] |
| Validation Platform | Flow Cytometry | Gold standard for cell proportion validation in complex mixtures | Requires >10% abundance for reliable detection [4] |
| Validation Platform | Synthetic Mixtures | In-silico created mixtures with known proportions | Enables precise spillover quantification [4] |
| Software Library | xCell2 R/Bioconductor | Primary implementation of xCell 2.0 algorithm | Includes pre-trained references for human and mouse [4] |
| Web Tool | xCell Web Application | User-friendly interface for enrichment analysis | No installation required, suitable for exploratory analysis [22] |
| Method Comparison | Deconvolution DREAM | Standardized benchmark for objective performance assessment | Independent validation dataset [4] |
Advanced Technical Implementation Details
Signature Generation Algorithm Enhancements
The xCell 2.0 signature generation process incorporates several key improvements over the original methodology. The algorithm employs an automated approach for handling cell type dependencies through ontological integration, extracting lineage information directly from the standardized Cell Ontology (CL) [4]. This eliminates the need for manual identification of lineage relationships, which was particularly challenging when dealing with custom references containing numerous cell types. The threshold criteria for gene inclusion has been modified to require that genes pass differential expression thresholds against at least 50% of cell types in the reference, providing adaptability to references with varying numbers of cell types [4].
Spillover Correction Mechanism
xCell 2.0 implements a sophisticated spillover correction system that uses in-silico simulated cell type mixtures to learn parameters modeling the linear relationship between signature enrichment scores and cell type proportions. The algorithm automatically selects the most transcriptionally distinct cell type as control for each target cell type, then calculates a spillover matrix reflecting pairwise spillover between all cell types (excluding those with lineage dependencies) [4]. The spillover correction strength (α) parameter allows users to balance between correcting genuine spillover effects and potential over-correction, with comprehensive validation identifying α=0.7 as optimal for most applications [4].
Clinical Application in Immunotherapy Response Prediction
In translational validation, xCell 2.0 was applied to bulk RNA-seq data from 2007 cancer patients prior to Immune Checkpoint Blockade (ICB) therapy across multiple cancer types. The xCell 2.0-derived tumor microenvironment features significantly improved prediction accuracy of patient response to ICB compared to models using only cancer type and treatment information [4]. Furthermore, xCell 2.0 outperformed other deconvolution methods and established prediction scores, demonstrating its potential for advancing precision medicine in cancer immunotherapy [4].
The accurate characterization of the tumor microenvironment (TME) is crucial for understanding cancer biology and developing effective therapies. Computational deconvolution algorithms like xCell enable researchers to infer cellular composition from bulk gene expression data, offering a scalable alternative to traditional methods like flow cytometry and immunohistochemistry (IHC) [48]. However, the clinical utility of these algorithms depends entirely on their validation against these established gold-standard techniques. This application note synthesizes evidence from multiple studies that have quantitatively correlated xCell predictions with cytometry and IHC data, providing researchers with a framework for assessing the algorithm's performance and limitations in different biological contexts.
Multiple independent studies have evaluated xCell's performance against conventional protein-based measurement technologies. The table below summarizes key validation findings across different disease contexts and sample types.
Table 1: Correlation of xCell Predictions with Cytometry and Immunohistochemistry Data
| Study Context | Validation Method | Key Correlated Cell Types | Correlation Strength | Reference |
|---|---|---|---|---|
| Rheumatoid Arthritis & Multiple Sclerosis (CLARITY) | Flow Cytometry (26 immune cell types) | ~50% of tested signatures | Strong correlation (r > 0.5) for ~50% of signatures; remainder showed moderate or no correlation [48]. | [48] |
| Acute Myeloid Leukemia (AML) | RT-qPCR (40 patient samples) | CD163 gene expression | Significant elevation in AML vs. controls (p < 0.001) [5]. | [5] |
| Acute Myeloid Leukemia (AML) | RT-qPCR (40 patient samples) | MRC1 gene expression | No significant differential expression [5]. | [5] |
| Triple-Negative Breast Cancer (TNBC) | Immunohistochemistry (SYSUCC cohort) | M2 Macrophages, CD8+ T cells, CD4+ memory T cells | Risk score based on xCell-predicted cells aligned with IHC and stratified patient survival (p < 0.05) [49]. | [49] |
Detailed Experimental Protocols
Protocol: Validating xCell Predictions Using Flow Cytometry Data
This protocol is adapted from a study that utilized the publicly available GSE93777 dataset to validate xCell and CIBERSORT outputs against extensive flow cytometry data [48].
I. Sample Preparation and Data Generation
- Sample Collection: Collect peripheral whole blood samples in appropriate anticoagulant tubes.
- Gene Expression Profiling: Isolate total RNA and perform gene expression analysis using a standardized platform, such as Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays or RNA sequencing. The same samples must be used for both deconvolution and flow cytometry.
- Flow Cytometry: For the matching samples, perform immune cell isolation and staining with a pre-defined panel of antibodies targeting 26 immune cell surface markers (e.g., CD19 for B cells, CD3 for T cells, CD4 for T-helper cells, CD8 for cytotoxic T cells). Analyze the stained cells using a flow cytometer to obtain absolute counts or relative fractions for each cell type.
II. Computational Analysis with xCell
- Data Preprocessing: Normalize the gene expression data according to standard procedures for the chosen platform.
- xCell Deconvolution:
- Input the normalized gene expression matrix into the xCell algorithm (R package v.1.1 or later).
- Run the analysis to obtain enrichment scores for 64 immune and stromal cell types.
- As per the xCell manual, exclude signatures representing cell types outside the bloodstream (e.g., chondrocytes, osteoblasts, fibroblasts) when working with blood samples [48].
III. Data Correlation and Validation
- Data Alignment: Match the xCell enrichment scores for specific immune cell types with the corresponding relative fractions obtained from flow cytometry.
- Statistical Analysis: Perform correlation analysis (e.g., Pearson or Spearman correlation) between the xCell scores and flow cytometry measurements for each cell type.
- Interpretation: A correlation coefficient (r) > 0.5 is generally considered evidence of a strong correlation, confirming the signature's validity for that specific cell type in the studied tissue [48].
Protocol: Validating xCell-Based Risk Models with Immunohistochemistry
This protocol outlines the process of validating a TME-based risk score, derived from xCell, using IHC on a patient cohort, as demonstrated in a TNBC study [49].
I. Cohort Selection and Model Construction
- Cohort Definition: Select a cohort of patients (e.g., with TNBC) with available gene expression data and clinical follow-up information. Divide into training and validation sets.
- xCell Analysis and Model Building:
- Process the training cohort's gene expression data with xCell to get enrichment scores for 64 cell types.
- Use univariate Cox regression to identify cell types with significant prognostic value.
- Input these prognostic cells into a Random Survival Forest (RSF) model to identify the most potent combination of cells for risk stratification (e.g., M2 macrophages, CD8+ T cells, CD4+ memory T cells) [49].
- Construct a final risk scoring system using multivariate Cox regression.
II. Immunohistochemical Validation
- Tissue Microarray (TMA) Construction: Use formalin-fixed, paraffin-embedded (FFPE) tumor tissue blocks from an independent validation cohort (e.g., the SYSUCC cohort). Construct TMAs.
- IHC Staining and Scoring:
- Cut TMA sections to 3-4 µm thickness.
- Perform antigen retrieval at high temperature (98°C) in citrate buffer (pH 6.0) for 10 minutes.
- Block endogenous peroxidase activity with 3% hydrogen peroxide.
- Incubate sections overnight at 4°C with primary antibodies against the key cell types identified by the model (e.g., anti-CD163 for M2 macrophages, anti-CD8 for CD8+ T cells).
- The next day, incubate with an HRP-conjugated secondary antibody for 30 minutes at room temperature.
- Develop the signal using a 3,3'-Diaminobenzidine (DAB) chromogen and counterstain with hematoxylin.
- Digital Pathology & Quantification: Scan the stained slides. The stained slides should be evaluated by two independent pathologists blinded to the clinical data. Quantify the density of positive cells in the tumor epithelium and stroma.
III. Clinical Correlation
- Risk Group Assignment: Use the IHC-based cell densities to assign patients into the same risk groups as defined by the xCell-based model.
- Survival Analysis: Perform Kaplan-Meier survival analysis and log-rank tests to confirm that the IHC-defined risk groups show significant divergence in overall survival, thereby validating the prognostic power of the computational model [49].
The Scientist's Toolkit
Table 2: Essential Research Reagents and Resources for xCell Validation
| Item | Function/Description | Example Use in Validation |
|---|---|---|
| Affymetrix GeneChip Human Genome U133 Plus 2.0 Array | Standardized microarray platform for gene expression profiling. | Generating bulk transcriptomic data from whole blood or tissue samples for xCell input [48]. |
| Flow Cytometry Antibody Cocktail | Panel of fluorescently-labeled antibodies against cell surface markers (e.g., CD3, CD19, CD4, CD8, CD56). | Quantifying true immune cell population fractions in matched samples for correlation with xCell scores [48]. |
| Primary Antibodies for IHC (e.g., anti-CD163, anti-CD8) | Antibodies for detecting specific cell types in FFPE tissue sections. | Validating the spatial localization and density of key cell types identified by xCell models [49]. |
| xCell R Package | Computational tool for estimating 64 immune and stromal cell type enrichments from gene expression data. | Digitally dissecting the TME to generate cell enrichment scores for downstream model building [5] [49] [50]. |
| Random Survival Forest (RSF) Model | Machine learning method for building prognostic models using survival data. | Identifying the most impactful combination of xCell-derived cell types for patient risk stratification [49]. |
Discussion and Technical Notes
The collective evidence indicates that xCell provides a reasonably accurate digital portrayal of the cellular TME, with approximately half of its signatures showing strong correlation with flow cytometry data [48]. However, performance is highly cell-type and context-dependent. For instance, while a risk model based on xCell-predicted M2 macrophages and T cells was validated with IHC in TNBC [49], the algorithm failed to correlate with RT-qPCR measurements of MRC1 in AML [5]. This underscores the critical importance of experimental validation for the specific cell types and disease models under investigation.
For optimal results, researchers should prioritize xCell signatures for broad immune cell lineages (e.g., CD8+ T cells, B cells) which generally show more robust performance, and should be cautious when interpreting results for closely related cell subsets or rare populations. When moving from discovery to clinical application, building a predictive model from xCell outputs and then validating that specific model with IHC on a independent cohort, as demonstrated in the TNBC study, provides a robust framework for translating computational findings into clinically actionable insights [49].
Immune checkpoint blockade (ICB) therapy has revolutionized cancer treatment, demonstrating durable remission across various cancer types. However, patient response is highly heterogeneous, with a significant proportion of patients failing to benefit from treatment. A major challenge in the field is the accurate prediction of which patients will respond to ICB therapy. The tumor microenvironment (TME) plays a crucial role in determining therapeutic outcomes, but its cellular complexity has been difficult to comprehensively characterize. xCell 2.0 addresses this challenge by providing robust digital dissection of the TME from bulk transcriptomic data, enabling superior prediction of ICB response compared to existing methods and clinical variables alone [16] [4].
This application note details how xCell 2.0, a signature-based algorithm for estimating cell type proportions from bulk gene expression data, significantly enhances ICB outcome forecasting. We present comprehensive performance benchmarks, detailed experimental protocols for applying xCell 2.0 to ICB response prediction, and essential computational tools for implementation.
Performance Benchmarking in ICB Response Prediction
Comparative Performance Against Established Methods
xCell 2.0 was rigorously evaluated using bulk RNA-seq data from 2,007 cancer patients across multiple cancer types collected prior to ICB treatment. The algorithm-derived TME features were compared to existing deconvolution methods and established prediction scores [16].
Table 1: Performance Comparison of xCell 2.0 in ICB Response Prediction
| Method Category | Specific Method/Model | Key Performance Metrics | Superiority of xCell 2.0 |
|---|---|---|---|
| Deconvolution Methods | 11 popular methods | Accuracy, consistency across biological contexts | Outperformed all 11 methods in benchmarking |
| Clinical Baseline Model | Cancer type + treatment information | Prediction accuracy | Significantly improved prediction accuracy |
| Established Prediction Scores | T-cell inflamed score, cytolytic activity score | Association with response | Derived features provided better prediction |
Technical Advantages Underlying Predictive Performance
The enhanced performance of xCell 2.0 in ICB response prediction stems from key algorithmic improvements:
- Minimized Spillover Effects: xCell 2.0 demonstrates the best performance in minimizing spillover effects between related cell types, crucial for accurate immune cell quantification [16]
- Automated Cell Type Dependency Handling: Integration of Cell Ontology (CL) automatically identifies lineage relationships, preventing biased comparisons between closely related cell types (e.g., T cells and CD4+ T cells) [16] [4]
- Robust Signature Generation: Modified threshold criteria requiring genes to be differentially expressed against at least 50% of reference cell types enhances signature reliability across diverse TME contexts [16]
Experimental Protocol for ICB Response Prediction
Sample Processing and Data Generation
Input Requirements:
- Bulk tumor tissue RNA-seq or microarray data from pre-treatment biopsies
- Minimum sample quality: RNA integrity number (RIN) > 7.0 recommended
- Standard gene expression quantification: TPM for RNA-seq, normalized intensities for microarrays
Data Preprocessing:
- Perform standard quality control of transcriptomic data
- Normalize using platform-appropriate methods (e.g., TMM for RNA-seq, quantile normalization for microarrays)
- Annotate genes to standard gene symbols matching xCell 2.0 reference
xCell 2.0 Analysis Workflow
Software Implementation:
Critical Parameters for ICB Prediction:
- Spillover correction strength (α): Use default value of 0.5 for balanced correction
- Signature specificity threshold: Maintain at 50% for robust TME characterization
- Immune cell focus: Prioritize CD8+ T cells, Tregs, macrophages, and dendritic cells for ICB response
Predictive Model Building
Feature Selection:
- Select xCell 2.0-derived immune cell abundances with highest variance across samples
- Include stromal cell populations (cancer-associated fibroblasts, endothelial cells)
- Calculate immune cell ratios (e.g., CD8+/Treg ratio) as additional features
Model Training:
- Employ machine learning classifiers (random forest or logistic regression recommended)
- Train on 70% of samples with stratified sampling by response status
- Validate on remaining 30% test set with multiple cross-validation iterations
Clinical Integration:
- Combine xCell 2.0 features with clinical variables (cancer type, prior treatments)
- Validate model on external datasets when available
Workflow Visualization
xCell 2.0 ICB Response Prediction Workflow
xCell 2.0 Algorithmic Improvements
Research Reagent Solutions
Table 2: Essential Research Tools for xCell 2.0 Implementation in ICB Studies
| Resource Category | Specific Tool/Resource | Function in ICB Response Prediction | Availability |
|---|---|---|---|
| Computational Tools | xCell 2.0 R/Bioconductor Package | Cell type deconvolution from bulk transcriptomic data | Bioconductor |
| Pre-trained References | Human Immune Cell Atlas (xCell2refs) | Reference signatures for 64 immune and stromal cell types | https://dviraran.github.io/xCell2refs |
| Validation Datasets | Deconvolution DREAM Challenge Dataset | Independent method validation and benchmarking | Publicly available |
| Clinical Data | ICB-treated Patient Cohorts (e.g., melanoma, NSCLC) | Model training and validation with response annotations | Controlled access |
| Bioinformatics Platforms | R Statistical Environment | Data analysis, visualization, and predictive modeling | Open source |
Technical Notes and Implementation Guidelines
Optimal Reference Selection for ICB Applications
For ICB response prediction studies, selection of appropriate pre-trained references is critical:
- Human Immune Cell References: Recommended for most ICB applications, providing comprehensive immune profiling
- Tumor Microenvironment-Specific References: Essential for solid tumors, capturing tissue-resident immune populations
- Custom Reference Generation: Advised for novel cancer types or specialized treatment contexts using scRNA-seq data
Quality Control Metrics
Implement these QC measures to ensure reliable predictions:
- Check for low abundance estimates across multiple cell types (may indicate poor sample quality)
- Verify expected immune cell presence (e.g., T cells in inflamed tumors)
- Confirm correlation between technical replicates >0.95
- Validate with known cell type markers when orthogonal data available
Interpretation Guidelines for ICB Response
Key xCell 2.0 features associated with improved ICB response:
- Elevated CD8+ T cell and memory T cell abundances
- Favorable immune cell ratios (e.g., CD8+/Treg ratio >2)
- Reduced immunosuppressive populations (M2 macrophages, specific myeloid subsets)
- Structured immune cell spatial patterns (when integrated with spatial transcriptomics)
The implementation of xCell 2.0 for ICB response prediction provides researchers and clinicians with a powerful tool for patient stratification, potentially enhancing therapeutic efficacy while minimizing unnecessary treatment and associated toxicities.
The tumor microenvironment (TME) has emerged as a critical determinant of cancer progression, therapeutic response, and patient outcomes across multiple malignancies. Real-world evidence (RWE) studies leveraging computational TME analysis are now providing unprecedented insights into cancer biology directly from clinical patient data. The xCell algorithm represents a cornerstone methodology in this field, enabling researchers to decipher cellular heterogeneity from bulk transcriptomic data of patient tumors. This gene signature-based method performs cell-type enrichment analysis to quantify the abundance of 64 immune and stromal cell types, creating comprehensive TME profiles that can be correlated with clinical outcomes [51] [10].
The latest iteration, xCell 2.0, introduces significant improvements for RWE applications, including automated handling of cell type dependencies and more robust signature generation. This enhanced algorithm demonstrates superior accuracy in benchmarking against other deconvolution methods and has proven particularly valuable for predicting response to immune checkpoint blockade therapy [4]. For drug development professionals, these methodologies offer a powerful approach to stratify patient populations, identify novel biomarkers, and understand mechanisms of treatment resistance using real-world transcriptomic data from clinical practice.
xCell Algorithm Methodology and Technical Protocols
Core Algorithm Principles and Workflow
The xCell algorithm operates through a multi-step process that transforms bulk gene expression data into enriched scores representing TME composition. The methodology employs single-sample Gene Set Enrichment Analysis (ssGSEA) to calculate enrichment scores for predefined gene signatures, then applies a spillover compensation technique to reduce dependencies between closely related cell types [10]. This technical approach integrates the advantages of gene set enrichment with deconvolution methods, allowing for robust quantification of diverse immune and stromal cell populations from standard transcriptomic datasets.
xCell 2.0 incorporates several key enhancements that improve its application in RWE studies. The updated version introduces a training function that permits utilization of any reference dataset, significantly expanding its flexibility for specific tissue types or experimental conditions. Additionally, it features automated handling of cell type dependencies through ontological integration, extracting cell type lineage information directly from the standardized Cell Ontology (CL) database. This automation eliminates the need for manual identification of lineage relationships, which was particularly challenging when dealing with custom references containing numerous cell types [4]. The algorithm also implements improved signature generation with modified threshold criteria that requires genes to pass expression thresholds against at least 50% of cell types in the reference, enhancing robustness across diverse reference datasets.
Practical Implementation Protocol
For researchers implementing xCell analysis in real-world studies, the following step-by-step protocol provides a standardized approach:
Table 1: Sample Data Requirements for xCell Analysis
| Data Type | Format Specifications | Preprocessing Needs | Quality Control |
|---|---|---|---|
| Bulk Tumor RNA-seq | TPM or FPKM normalized | Batch effect correction | RIN >7 recommended |
| Microarray Data | Normalized expression values | Combat algorithm for multi-study integration | Present call rates >95% |
| Single-cell RNA-seq | Count matrices | Standard Seurat workflow | Mitochondrial reads <20% |
| Clinical Outcomes | Structured data (CSV/TSV) | Censoring handling for survival | Follow-up duration documentation |
Step 1: Data Preparation and Normalization Begin with raw gene expression data (microarray or RNA-seq) from tumor samples. For RNA-seq data, convert to TPM or FPKM normalized values. For multi-study integrations, apply batch effect correction using established methods like the ComBat algorithm [52]. Ensure data quality with standard metrics including RNA Integrity Number (RIN) >7 for RNA-seq or present call rates >95% for microarray data.
Step 2: xCell Score Calculation Execute xCell analysis using the available R package or web application (https://xcell.ucsf.edu/). For standard analyses, use the pre-trained references provided by the developers. For specialized applications (e.g., specific cancer types or treatment contexts), consider developing custom reference objects using the xCell 2.0 training function [4]. Run the algorithm with 1000 permutations and apply quantile normalization as recommended for optimal performance.
Step 3: Spillover Compensation and Score Transformation Apply the built-in spillover correction to minimize cross-contamination between related cell types. The algorithm uses in-silico simulated cell type mixtures to learn parameters that model the linear relationship between signatures' enrichment scores and cell type proportions, with automatic selection of control cell types as the most distinct cell type according to gene expression correlation [4].
Step 4: Data Integration and Statistical Analysis Integrate the resulting xCell scores with clinical and molecular data for correlation analysis, survival modeling, or treatment response assessment. Employ appropriate multiple testing correction for the high-dimensional cell type data, such as Benjamini-Hochberg false discovery rate adjustment.
Breast Cancer Applications
TME-Based Risk Stratification and Prognostication
In breast cancer research, xCell-driven TME analysis has enabled refined patient stratification beyond conventional classification systems. A seminal RWE study analyzed 1,901 breast cancer patients from the Bernard cohort, applying xCell algorithm to quantify 64 immune and stromal cell types in the TME [51]. Unsupervised clustering of these TME profiles revealed three distinct microenvironment clusters with significant survival differences (log-rank test, p=0.006). This TME-based classification provided prognostic value independent of standard clinicopathological factors, demonstrating the clinical utility of computational TME analysis.
Further investigation employed multiple machine learning algorithms to develop a novel immune-related pathway-based risk score (IPRS) based on TME characteristics. This nine-pathway signature stratified patients into IPRS-high and IPRS-low groups with markedly different overall survival (log-rank test, p<0.0001) [51]. Multivariate analysis confirmed IPRS as an independent prognostic biomarker after adjustment for standard clinicopathologic characteristics, including subtype, ER status, HER2 status, PR status, grade, tumor size, and tumor stage. The IPRS-low group exhibited characteristic TME features including increased immune-related scores (cytolytic activity, MHC expression, T cell-inflamed gene expression profile), elevated ESTIMATE immune and stromal scores, and decreased tumor purity, suggesting these patients harbor a more robust anti-tumor immune response.
Triple-Negative Breast Cancer Microenvironment Profiling
In triple-negative breast cancer (TNBC), where therapeutic options remain limited, xCell analysis has revealed distinct TME patterns with clinical implications. A comprehensive analysis of 158 TNBC samples from The Cancer Genome Atlas identified six tumor-infiltrating immune cells with significant prognostic value through univariate Cox regression [10]. Random survival forest modeling further refined these to three key cell types: M2 macrophages, CD8+ T cells, and CD4+ memory T cells.
Table 2: xCell Applications in Breast Cancer Studies
| Study Focus | Cohort Size | Key TME Findings | Clinical Implications |
|---|---|---|---|
| General BC Classification | 1,901 patients | 3 TME clusters with distinct survival | Independent prognostic value beyond standard staging |
| TNBC Microenvironment | 158 patients (TCGA) | M2 macrophages, CD8+ T cells, CD4+ memory T cells drive prognosis | Risk scoring system identifies immunotherapy candidates |
| TNBC Validation | 297 patients (METABRIC) | 4 immunophenotypes with differential survival | High-risk group shows enriched immune checkpoint molecules |
| Pathway Analysis | 4 independent cohorts | 9-pathway IPRS signature stratifies survival | IPRS-low shows enhanced immune activity and better outcome |
Based on these determinants, researchers developed a risk scoring system that categorized TNBC patients into four distinct immunophenotypes: M2low, M2highCD8highCD4high, M2highCD8highCD4low, and M2highCD8low [10]. When merged into low-risk (types 1-2) and high-risk (types 3-4) groups, significant survival differences emerged that were subsequently validated in independent cohorts (METABRIC, n=297; GSE58812, n=107). The low-risk group demonstrated superior survival outcomes and characteristic TME features including enriched immune-related pathways and elevated expression of immune checkpoint molecules (PD-L1, PD-1, CTLA-4), suggesting these patients might derive particular benefit from immunotherapy approaches.
Colorectal Cancer Applications
Metastatic CRC and Tumor Microenvironment Dynamics
Colorectal cancer studies have leveraged xCell algorithm to investigate TME changes associated with metastasis and therapy resistance. A comprehensive bioinformatics analysis of metastatic CRC (mCRC) dissected transcriptomic data from TCGA and GEO repositories, employing xCell to characterize immune infiltration patterns [41]. This approach identified 7 tumor-infiltrating immune cell subtypes with significant abundance differences between metastatic and non-metastatic colorectal cancer cohorts. Integrative analysis further revealed 28 immune-related metastatic colorectal cancer differentially expressed genes (ICDEGs) in metastatic lesions, with 9 pivotal hub genes (AGTR1, CD86, CMKLR1, FGF1, FYN, IL10RA, INHBA, TNFSF13B, and VEGFC) emerging as potential diagnostic biomarkers for mCRC.
The relationship between Consensus Molecular Subtypes (CMS) and TME characteristics in CRC was further elucidated through xCell analysis of 765 primary CRC samples and 442 metastasis samples [52]. This investigation revealed that 64% of CRC metastases exhibited concordant CMS groups with matched primary tumors, and the TME of metastases maintained similarity to that of primary lesions. However, organ-specific patterns emerged: liver metastases were predominantly CMS2, while lung and peritoneal metastases were mainly CMS4, supporting the "seed and soil" hypothesis that tumor cells of different molecular subtypes show preferential metastasis to specific organs. xCell analysis further quantified distinct immune-stromal infiltration patterns across metastatic sites, with liver metastases showing reduced cancer-associated fibroblasts (CAFs), lung metastases displaying increased CD4+ T cells and M2-like macrophages, and peritoneal metastases exhibiting elevated M2-like macrophages and CAFs compared to primary tumors.
ADCP-Related Gene Signatures and Prognostic Modeling
Another innovative application of xCell in CRC research explored antibody-dependent cellular phagocytosis (ADCP) mechanisms within the TME. Researchers established a prognostic model based on 7 ADCP-related genes using TCGA-CRC cohort data [53]. xCell algorithm was employed to analyze the TME of high- and low-ADCP-related risk score (ADCPRS) groups, revealing distinct immune landscapes. Single-cell RNA sequencing data validation (GSE178341) confirmed expression of the 7 feature genes across 8 cell clusters (Monocytes, CD8+ T cells, Epithelial cells, B cells, Macrophages, HSC, Endothelial cells, and Fibroblasts), with AUCell scoring showing higher scores predominantly in B cells and macrophages [53].
The ADCPRS groups demonstrated significantly different immune infiltration patterns, with Th1 cells, iDCs, and Th2 cells showing higher abundance in the low-ADCPRS group. This comprehensive analysis integrated TME features with somatic mutation profiling, revealing high mutation rates in both groups with APC and TP53 as the top two most frequently mutated genes. The study further connected these TME findings to therapeutic implications through drug sensitivity analysis, identifying Dasatinib, Benzaldehyde, and Tegafur as potential therapeutic agents for CRC patients based on their TME profiles [53].
Ovarian Cancer Applications
Spatial Heterogeneity in the Ovarian Cancer Microenvironment
Ovarian cancer presents particularly complex TME heterogeneity that influences therapy response and patient outcomes. A landmark spatial transcriptomics study analyzed 272,389 CD45+ immune cells from 111 tumor and non-malignant tissue samples across tubo-ovarian, endometrial, and cervical cancers [54]. This comprehensive single-cell atlas identified extensive immune cell diversity, including 11 distinct subsets of monocytes/macrophages, 6 CD4+ T cell subsets, 8 CD8+ T cell subsets, and 5 B cell subsets, each with unique distribution patterns and functional characteristics.
The investigation revealed clinically relevant macrophage subpopulations with opposing prognostic implications. A pro-angiogenic macrophage subset driven by NF-κB signaling was associated with worse clinical outcomes, while an interferon-primed macrophage subset correlated with improved survival by recruiting T cells through CXCL9/10/11 secretion [54]. These findings were validated through multi-color immunohistochemistry, confirming the functional significance of these distinct macrophage populations in the ovarian cancer TME. T cell analysis further demonstrated dynamic roles in tubo-ovarian cancer, with CD8 exhausted T cells (Tex) contributing to immune dysfunction and poor prognosis, while CD8 tissue-resident memory T cells (Trm) in early-stage tumors supported immune surveillance.
Immune Phenotypes and Immunotherapy Implications
Another spatial profiling study of ovarian cancer microenvironment characterized regions of spatially distinct TIME phenotypes to assess whether immune infiltration patterns predict presence of immuno-oncology targets [55]. Using Digital Spatial Profiling combined with image analysis, researchers classified TIME phenotypes into three categories: diffuse immune infiltration, focal immune niches, and immune exclusion. Tumors with diffuse immune infiltration showed increased tumor-immune spatial interactions and higher expression of immunotherapy targets including IDO1, PD-L1, PD-1 and Tim-3, while focal immune niches contained more CD163+ macrophages and demonstrated a preliminary association with worse outcome.
This study further revealed histotype-specific TME patterns with therapeutic implications. High-grade serous OC showed an overall stronger immune response and presence of multiple targetable checkpoints, while low-grade serous OC was associated with diffuse infiltration and high expression of STING [55]. Endometrioid OC had higher presence of CTLA-4, whereas mucinous and clear cell OC were dominated by focal immune clusters and immune-excluded regions. Importantly, immune exclusion was associated with presence of Tregs and Fibronectin, suggesting potential mechanisms of immune evasion that might be therapeutically targeted.
Experimental Protocols and Reagent Solutions
Integrated xCell Analysis Protocol for Multi-Cancer Studies
For researchers implementing xCell analysis across multiple cancer types, the following standardized protocol ensures consistent and reproducible results:
Step 1: Data Collection and Quality Control
- Obtain bulk gene expression data from tumor samples (RNA-seq or microarray)
- For multi-study integrations, apply batch effect correction using ComBat algorithm
- Verify data quality metrics: RIN >7 for RNA-seq, present call rates >95% for microarrays
- Collect matched clinical data including survival, treatment history, and response outcomes
Step 2: xCell Implementation and Customization
- Execute xCell analysis using R package or web interface (https://xcell.ucsf.edu/)
- For standard analyses, use pre-trained references provided by developers
- For cancer-specific applications, consider xCell 2.0's training function with custom references
- Run algorithm with 1000 permutations and apply quantile normalization
Step 3: TME Phenotype Classification
- Perform unsupervised clustering (e.g., consensus clustering) on xCell output
- Identify robust TME clusters using stability metrics and biological coherence
- Correlate TME clusters with clinical and molecular variables
- Validate findings in independent cohorts when available
Step 4: Integration with Multi-Omics Data
- Combine xCell results with mutation, copy number, and epigenetic data
- Perform pathway analysis on TME-defined patient subgroups
- Investigate spatial relationships through integration with imaging data when available
- Correlate specific immune cell populations with treatment responses
Step 5: Validation and Functional Confirmation
- Validate computational findings using orthogonal methods (IHC, flow cytometry)
- Perform single-cell RNA sequencing validation on subset of samples
- Conduct in vitro or in vivo experiments to confirm functional mechanisms
- Develop clinical assay prototypes for translational application
Essential Research Reagent Solutions
Table 3: Key Research Reagents for TME Studies
| Reagent/Category | Specific Examples | Research Application | Considerations |
|---|---|---|---|
| Transcriptomic Profiling | RNA-seq kits, Microarray platforms | Bulk gene expression for xCell input | RNA quality critical for reliable results |
| Single-cell RNA-seq | 10X Genomics Chromium, Smart-seq2 | Validation of xCell predictions, rare cell identification | Higher resolution but costlier than bulk |
| Spatial Transcriptomics | GeoMx Digital Spatial Profiler, Visium | Spatial context for TME patterns | Preserves tissue architecture information |
| Immunohistochemistry | Validated antibodies for immune markers | Protein-level validation of cell abundances | Quantitative image analysis recommended |
| Cell Line Models | Primary cell cultures, organoids | Functional validation of TME interactions | Better represents human biology than immortalized lines |
| Computational Tools | xCell R package, ESTIMATE, CIBERSORTx | TME deconvolution and analysis | Parameter optimization needed for specific contexts |
The integration of xCell algorithm with real-world evidence has fundamentally advanced our understanding of tumor microenvironment complexity across breast, colorectal, and ovarian cancers. These computational approaches have revealed clinically meaningful patient subgroups, identified novel therapeutic targets, and provided insights into treatment resistance mechanisms. The development of xCell 2.0 with enhanced flexibility and accuracy promises to further accelerate these discoveries through improved handling of cell type dependencies and more robust signature generation [4].
Future applications of TME analysis in real-world evidence will likely focus on several key areas: predictive biomarker development for immunotherapy response, understanding TME evolution during treatment, and integrating multi-omics data for comprehensive microenvironment profiling. Additionally, the growing availability of spatial transcriptomics technologies will provide critical validation of computational predictions and reveal new dimensions of cellular organization within tumors [55]. As these methodologies continue to mature, they hold significant potential to guide personalized treatment strategies and improve outcomes for cancer patients across diverse malignancies.
For the research community, ongoing development of standardized protocols, shared reference datasets, and validated computational pipelines will be essential to maximize the translational impact of these approaches. The integration of TME analysis into prospective clinical trials represents a particularly promising direction, potentially enabling real-time patient stratification and biomarker-driven treatment assignment based on comprehensive microenvironment characterization.
Conclusion
xCell 2.0 represents a significant advancement in digital cytometry, offering researchers a robust, flexible, and validated framework for TME deconvolution that maintains high performance across diverse biological contexts and reference types. The algorithm's ability to accurately predict response to immune checkpoint blockade and other therapies underscores its potential as a biomarker development tool for precision oncology. Future directions should focus on expanding reference atlases for specific cancer types, integrating multi-modal data including epigenomic and spatial information, and validating clinical utility in prospective trials. As TME-directed therapies continue to evolve, xCell and similar computational tools will play an increasingly critical role in unlocking the therapeutic potential of the tumor microenvironment, ultimately enabling more personalized and effective cancer treatments.
References
- 1. Pan-cancer immune and stromal deconvolution predicts ... [https://www.nature.com/articles/s41598-025-09075-y]
- 2. Pan-cancer immune and stromal deconvolution predicts ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12227561/]
- 3. Community assessment of methods to deconvolve cellular ... [https://www.nature.com/articles/s41467-024-50618-0]
- 4. xCell 2.0: robust algorithm for cell type proportion estimation ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03784-3]
- 5. Constructing a tumor immune microenvironment-driven ... [https://www.nature.com/articles/s41598-025-03557-9]
- 6. Benchmark of cellular deconvolution methods using a multi ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03552-3]
- 7. Computational pathology annotation enhances the resolution ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12420830/]
- 8. Enhanced prediction of breast cancer patient response to ... [https://www.sciencedirect.com/science/article/abs/pii/S0304383525006731]
- 9. Immunoinformatic analysis of role of tumor ... [https://www.sciencedirect.com/science/article/pii/S3050787125000162]
- 10. A risk scoring system based on tumor microenvironment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9021102/]
- 11. xCell 2.0: robust algorithm for cell type proportion ... [https://pubmed.ncbi.nlm.nih.gov/41044684/]
- 12. xCell 2.0: Cell Type Enrichment Analysis [https://aran-lab.com/xcell2-vignette]
- 13. Performance of tumour microenvironment deconvolution ... [https://www.nature.com/articles/s41467-023-41385-5]
- 14. Multiomics integration analysis identifies tumor cell-derived ... [https://pubmed.ncbi.nlm.nih.gov/40846326/]
- 15. xCell: digitally portraying the tissue cellular heterogeneity ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1349-1]
- 16. xCell 2.0: robust algorithm for cell type proportion estimation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492622/]
- 17. A Novel Immune-Related Gene Signature to Identify the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9351622/]
- 18. Comprehensive Analysis of Tumor Microenvironment ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2021.616073/full]
- 19. Genomic investigation of innate sensing pathways in the ... [https://bmccancer.biomedcentral.com/articles/10.1186/s12885-024-12944-w]
- 20. Introduction to Linear transformation and application in ... [https://medium.com/@yokeswaran1718/introduction-to-linear-transformation-and-application-in-data-science-151f7009663c]
- 21. Cellular heterogeneity map of diverse immune and stromal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7357563/]
- 22. xCell [https://aran-lab.com/software/xcell/]
- 23. Comprehensive Analysis Identifies and Validates the ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.838374/full]
- 24. Expression Matrix [https://docs.kbase.us/data/upload-download-guide/expression-matrix]
- 25. Generating a Gene Expression Matrix [https://ucdavis-bioinformatics-training.github.io/2022-December-Spatial-Transcriptomics/data_reduction/Expression_Matrix]
- 26. dviraran/xCell: Cell types enrichment analysis [https://github.com/dviraran/xCell]
- 27. Comparison of Vendor-Pretrained and Custom-Trained Deep ... [https://pubmed.ncbi.nlm.nih.gov/39767212/]
- 28. xCell2 (development version) [https://www.bioconductor.org/packages/devel/bioc/html/xCell2.html]
- 29. Molecular Profiling and Tumor Microenvironment in ... [https://pubmed.ncbi.nlm.nih.gov/40387017/]
- 30. A novel tumor microenvironment-related gene signature for ... [https://www.nature.com/articles/s41598-025-25463-w]
- 31. The microenvironment cell index is a novel indicator for the ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-05950-w]
- 32. A breast cancer classification and immune landscape ... [https://www.nature.com/articles/s41698-023-00482-w]
- 33. Exploring the tumor microenvironment of breast cancer to ... [https://www.nature.com/articles/s41598-025-97784-9]
- 34. Single-cell and spatial transcriptomics integration [https://pmc.ncbi.nlm.nih.gov/articles/PMC12528096/]
- 35. Integrating single-cell and spatial transcriptomics to elucidate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9888017/]
- 36. Integrating spatial and single-cell transcriptomics data ... [https://www.nature.com/articles/s41467-023-43629-w]
- 37. Integrating spatial and single-cell transcriptomics reveals ... [https://www.nature.com/articles/s41419-024-06598-6]
- 38. Integrated multi-omics analysis of single-cell and spatial ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1612623/full]
- 39. A protocol for acquiring high-quality single-cell multi-omics ... [https://www.sciencedirect.com/science/article/pii/S2666166724005951]
- 40. Bioconductor - xCell (development version) 2 [http://bioconductor.unipi.it/packages/3.19/bioc/html/xCell2.html]
- 41. Exploration of effective biomarkers and infiltrating immune ... [https://pubmed.ncbi.nlm.nih.gov/41006647/]
- 42. Bulk vs. single cell RNA-seq: Experimental differences and ... [https://www.10xgenomics.com/blog/bulk-vs-single-cell-RNA-seq-experimental-differences-and-advantages-of-single-cell-resolution]
- 43. xCell: digitally portraying the tissue cellular heterogeneity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5688663/]
- 44. Navigating single-cell RNA-sequencing: protocols, tools ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12085826/]
- 45. From bulk, single-cell to spatial RNA sequencing [https://www.nature.com/articles/s41368-021-00146-0]
- 46. A Beginner's Guide to Analysis of RNA Sequencing Data [https://pmc.ncbi.nlm.nih.gov/articles/PMC6096346/]
- 47. Benchmarking of cell type deconvolution pipelines for ... [https://www.nature.com/articles/s41467-020-19015-1]
- 48. Revealing the immune cell subtype reconstitution profile in ... [https://www.nature.com/articles/s41598-023-34384-5]
- 49. A risk scoring system based on tumor microenvironment cells to predict prognosis and immune activity in triple-negative breast cancer | Breast Cancer [https://link.springer.com/article/10.1007/s12282-021-01326-w]
- 50. Identification and validation of tumor microenvironment-related prognostic biomarkers in breast cancer - Zhong - Translational Cancer Research [https://tcr.amegroups.org/article/view/55919/html]
- 51. Tumor Microenvironment Characterization in Breast ... [https://www.mdpi.com/2073-4425/13/11/1976]
- 52. Exploring features between primary and metastases of CRC | CMAR [https://www.dovepress.com/seed-and-soil-consensus-molecular-subgroups-cms-and-tumor-microenviron-peer-reviewed-fulltext-article-CMAR]
- 53. Revelation of prognosis and tumor of microenvironment ... colorectal [https://clinicalproteomicsjournal.biomedcentral.com/articles/10.1186/s12014-025-09553-5]
- 54. Single‐cell landscape of the tumour immune ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12640613/]
- 55. Spatial tumor immune microenvironment phenotypes in ... [https://www.nature.com/articles/s41698-024-00640-8]